
Abstract
During Iraq-Iran conflict, chemical weapons, particularly SM gas, were used numerous times, whose aftereffects are still present. This study aimed to compare serum proteome in the chronic ML (n = 10) and HC (n = 10). TMT label-based quantitative proteomics was used to examine serums from two groups. Among total significant proteins, 14 proteins were upregulated (log2 ≥ FC 0.5, p 0.05), and 6 proteins were downregulated (log2 ≤ FC − 0.5, p 0.05). By helping PPI network, and EA, 11 main pathways connected to significantly different protein expression levels were discovered, including inflammatory and cell adhesion signaling pathways. It may be deduced that the wounded organs of exposed individuals experience poor repair cycles of cell degeneration and regeneration because certain repair signals were elevated while other structural and adhesion molecules were downregulated. The systems biology approach can help enhance our basic knowledge of biological processes, and contribute to a deeper understanding of pathophysiological mechanisms, as well as the identification of potential biomarkers of disease.
Similar content being viewed by others


Introduction
Sulfur mustard (SM), bis (2-chloroethyl) sulfide, known as "mustard gas", is a type of chemical warfare agent with destructive effects on the lung, eye, skin, cardiovascular and digestive systems of humans [1, 2]. The lung is one of the body's organs that is vulnerable to injury. There is evidence that SM may produce a number of lung problems, including sub-epithelial fibrosis, chronic bronchitis, and chronic pulmonary obstruction, around 40 years after exposure (COPD). Patients exposed to SM have clinical symptoms that are comparable to those of COPD. Depending on the severity of pulmonary complications caused by SM, the patient’s exposure to SM is divided into three categories: mild, moderate, and severe [3]. Considering the uncertainty of exact pathophysiology of lung exposed to SM, several mechanisms in the lungs were proposed, including oxidant-antioxidant imbalance, immune system irregularities (systemic innate and adaptive immune cell alterations including: NK cells, CD4+ and CD8+ T cells) and increased inflammatory mediators, such as cytokines in the lung tissue, Broncho alveolar lavage (BAL), as well as serum [4,5,6,7,8,9,10,11]. Recent studies on the immunopathogenesis of chronic lung diseases, such as COPD, which is very similar to the disease under study, have shown an association with changes in Th17/Treg [12, 13].
Unfortunately, the exact mechanism of the effects of SM toxicity on the lungs over a long period of time is not known. Consequently, based on the explanations of the clinical symptoms of these patients, it is required to conduct more comprehensive research to know more about the symptoms of the mustard lung (ML) in the chronic phase of the disease. Treatment and diagnosis of people with ML, compared to healthy controls (HC) with a systems biology approach, are helpful in more accurate diagnosis of this disease. Regarding the systemic nature of this disease, to better understand the biological mechanisms of the disease, systems biology approach is needed. Proteomics studies, especially labeling method by Tandem Mass Tag® (TMT®) coupled with LC–MS/MS technology, are one of the best choices to identify and quantify the proteins in ML and HC groups which can be used in serum samples [14,15,16]. After the filtration of the proteome data and obtaining the differential abundance proteins (DAPs), the specific candidate proteins of this disease were identified to analyze the data resulting from the proteomics study. Based on enrichment analysis (EA), along with other bioinformatics analyses for protein identification and quantification candidates. EA is a powerful tool to examine the relationship between phenotype disease and a group of genes, as well as proteins. The selection and input criteria for EA are based on quantitative expression data for each gene or protein [17].
Gene ontology (GO) deals with the characteristics and functions of genes and genetic products. GO generally covers three domains (cellular component, molecular function, and biological process) [18]. The coordination of intracellular mechanisms is in terms of molecular interactions. Molecular interactions, which are continuous, cause the molecular pathways of cells to regulate and execute complex processes [18]. Among biological samples, the serum is a part of the blood which contains proteins, and other biological molecules which represent the body's biological mechanisms which is rich in biological information; using blood as the source of biomarkers can be a less invasive and less expensive method [19,20,21,22]. Nowadays, the biomarkers can be obtained via technologies, such as proteomics [23,24,25]. This study aimed to find proteome patterns in chronic ML. The reason to choose men is that the mechanism of SM effect on women may have different pathophysiology [26, 27], and due to the grouping of people included and the lack of access to the statistical population of women. In this regard, via systems biology approach, this study can be very useful to enhance our basic knowledge of biological processes and a deeper understanding of pathophysiological mechanisms of disease, as well as in clinical research to identify potential biomarkers of disease plus drug targets.
Materials and methods
Study population and design
This study was conducted with an ethical code at Baqiyatallah University of Medical Sciences (IR.BMSU.REC.1395.381). The participants were categorized due to degree of lung disease before being sampled based on American Thoracic Society (ATS) categorization. All volunteers were asked to participate in routine clinical tests, such as pulmonary function testing (PFT) (Multi-Functional Spirometer HI-801), carbon monoxide (CO) ( Bedfont piCO™ Smokerlyzer®), as well as hematological (Mindray Bc-3000) and biochemical analysis (Mindray Bs-230) consisting of CBC diff, FBS, lipid profile, Uric acid, AST, ALT Urea, Creatinine, and CRPq. Adult men with ML (n = 10) and HC (n = 10) took part. The inclusion and exclusion criteria of ML and HC groups are mentioned in Table 1. ML and HC would be able to participate in the research once the inclusion and exclusion criteria were checked and adapted for them. The final confirmation of ML disease was done by a specialist in the medical committee on verifying chemical-warfare damaged people. According to committee’s opinion, ML and HC participants were chosen. Schematic representation of study design is presented in Fig. 1.

Schematic representation of the study design, proteomic work flow and bioinformatics analysis
Sample processing
Sample preparation
Blood samples were taken from these subjects to perform the mentioned tests. For this purpose, after filling out the questionnaire and obtaining informed consent, 10 ml of human's whole blood samples from ML and HC groups, which were taken based on age and gender in the Baqiyatallah Hospital. Then, between 8 and 9 o'clock, the blood sample of ML and HC participants was collected as 12-h fasting in a blood clot tube without anticoagulant (BD Vacutainer® Venous Blood Collection). Then, the blood was kept at room temperature (RT) for 10 min, and after clotting, to separate the serum, the samples were centrifuged with a refrigerated centrifuge (2500 g for 10 min) (Sigma 3-30KS). The serums were aliquoted into polyethylene tubes (Eppendorf Tubes®1.5 ml). Then, the serum samples were lyophilized (4 °C for 24 h). The samples were kept in a deep freezer at 80 °C (Thermo Scientific™) until they were used.
Lyophilized samples for proteomics analysis were sent to Australian Proteome Analysis Facility (APAF) laboratory. In brief, lyophilized sample pellets were reconstituted to deplete abundant proteins and washed in phosphate buffer saline buffer (PBS). The samples were subjected to the abundant protein depletion kit (CAT NO: 85165 Thermo Scientific Pierce™). Using protein assay (CAT NO: 23235 Thermo Scientific™), the concentration of proteins in the lyophilized serum samples was determined. Using Dithiothreitol (DTT) and Ioadoacetamid (IAA), disulfide bonds in cysteines were reduced, and their number diminished. Eventually, using trypsin enzyme, the proteins were digested for the sequencing stage.
LC–MS/MS analysis
Isobaric Label Reagent TMT-10plex (CAT NO: 90110 Thermo Scientific™) peptide labeling was done based on the manufacturer's instructions for overall procedure. In brief, anhydrous acetonitrile (ACN), hydroxylamine (5%) was used to label each protein sample at RT. A "label check" experiment was performed before pooling the samples to ensure an equal amount of total peptides were pooled from all samples. TMT-labeled peptide samples were pooled at a 1:1 ratio across all samples and vacuum dried after determining the normalization factor. Desalting using C18 solid-phase extraction and vacuum centrifugation to full dryness was used to clean the samples. In the next stage, the samples were fractionated using a high pH reversed-phase kit (CAT NO: 84868 Thermo Scientific™). At this stage, peptides and proteins were separated using a nanoLC system via a nano-LC column. (Halo-C18, 160 Å, 2.7 µm, 100 µm × 20 cm). For MS/MS experiments, the eluent from the column was pumped into the mass spectrometer's ionization chamber (Thermo Scientific™—Q Exactive HF-X™ Hybrid Quadrupole—Orbitrap Mass Spectrometer). Peptide precursors with molecular weights ranging from 350 to 1850 m/z were scanned at 60 k resolution. Higher Energy Collisional Dissociation (HCD) was used to fragment the 20 most energetic ions in the survey scan, with a normalized collision energy of 35 and a precursor isolation of 0.7 m/z. Dynamic exclusion was set to 90 s, and MS/MS scan resolution was set to 60 k.
Data analysis
Proteome Discoverer™ software (version 2.1, Thermo Fisher Scientific™) was used to search the raw data files for each sample set for data analysis of Orbitrap Fusion data. The data were compared to all Homo sapiens sequences retrieved from SwissProt database using the search engines SequestHT (version 2018) and Mascot (version 2.4, Matrix Science, London, UK) (version 2018). The names of proteins were determined, and the gene symbol was obtained from the Uniprot database (http://www.uniprot.org), which contains 95,106 human proteins, including isoforms and unreviewed (Homo sapiens). Table 2 reports the parameters for data processing. Using the abundances of the sample controls as the denominators, TMT-10 plex kit calculated the quantitative ratios in two sets.
Differential abundance proteins (DAPs) analysis
A linear model was used for the statistical analysis of differentially abundant proteins using the Limma package, which is a fundamental component of Bioconductor in the R programming environment [28, 29]. Using Limma Package (|Log2FC|> 0.5, t-test p value < 0.05) to find DAPs, a comparison among the data from proteome sequence analysis was done. The normalized protein areas between control and disease samples were compared using Limma Package (|Log2FC|> 0.5, t-test p value < 0.05) in serum samples.
Network and enrichment analysis (EA)
Physical relationships among differentially abundant proteins were searched for protein–protein interactions (PPIs) using GeneMANIA tool (https://genemania.org/) to uncover more effective proteins in ML patients compared to HC PPIs network [30].
A famous database, Functional Enrichment analysis tool (FunRich) [31, 32], was used to determine how enriched biological pathways (KEGG) and GO Enrichment in patients compared to controls were related to each other [33, 34]. GO Consortium and KEGG pathway analysis provide functional annotations that support high-throughput data such as proteomes with a system biology approach.
Results
Study participants
A total of ten ML and ten HC were recruited. Males made up the entire group of 20 subjects. For ML, the average age was 53.7 years (range 48–60), while for HC, the average age was 47.5 years (range 45–55). Among the participants in the study, people with a BMI < 30.0 were included in the study. Height, weight, BMI index, spirometry indices, and leukocyte count did not significantly vary between the two groups. Only the difference in HRCT values between the two groups among the clinical cases presented in Table 3 was statistically significant; thus, all participants in the ML group had an HRCT value more than 8.0, whereas none in the HC group did. We could reach 20 ML and HC groups out of the 56 participants who were recruited. Serum was taken from the remaining participants and made accessible for proteomic analysis. These descriptive statistics are presented in Table 3.
Proteomic characteristics and identification of DAPs
The high abundant proteins decreased the fibrinogen, 1-acid glycoprotein, 1-antitrypsin, haptoglobin, 2-macroglubulin, IgA, albumin, IgG, apolipoprotein A-I, IgM, apolipoprotein A-II, and transferrin. To study differences in the expression in ML and HC serum samples, TMT labeling LC–MS/MS proteomic approach was used.
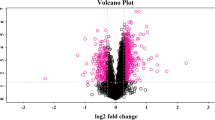
Based on the sequencing data of the proteome of ML and HC serum sample to find DAPs, overall, 20 proteins were found with significant differential protein expressions. P values were calculated from criteria t-test statistics (p value < 0.05), (|Log2FC|> 0.5) in serum samples, where log2FC ≥ 0.5 was upregulated, while log2FC ≤ -0.5 was downregulated. A total of 20 proteins for ML versus HC, including 14 upregulated and 6 downregulated proteins (Tables 4, 5), were found as shown in the volcano diagram (Fig. 2). Furthermore, the number of significant and non-significant proteins is given in Additional file 1.

Volcano diagram for significant expression differences proteins of ML versus HC
Gene ontology (GO) and pathway analysis
The unweighted PPI network was constructed using GeneMANIA tool by 20 proteins for assay GO enrichment analysis (up and down) (Fig. 3). Furthermore, more explanation details about the PPI network were provided in Additional file 2. GO enrichment analysis in the cellular component showed that the proteins were massively enriched in extracellular and exosomes (56.3%), followed by in the cytoskeleton (31.3%). In molecular function analysis, the largest proportions of proteins were involved in receptor activity (17.6%) and in cytoskeletal protein binding, and complement activity (11.8%). In biological process analysis, proteins were involved in response to cell growth (23.5%), immune response (17.6%), and protein folding, peptide metabolism (5.9%) (Fig. 4). Additionally, KEGG pathway enrichment analysis revealed a total of 11 primary pathways that were strongly connected to proteins with various expressions (Table 6).

PPI network. The analysis included proteins that showed a significantly differential abundance between ML and HC groups. Proteins are represented as nodes while interactions appear as link. The quantity of link relates to the strength of the interaction associations (In the colors used in the figure, there is a color gradient from red to green that red color was used to high degree proteins in the PPI, green color was used to low degree proteins in the PPI)

GO enrichment analysis in BP, CC and MF. Note: A refers to biological process, B refers to cellular component, C refers to molecular function
Discussion
During any type of disease, the proteins are the most functional and effective molecules. Understanding protein relationships gives insight into previously unknown aspects of the pathophysiology of hazardous substance interactions with human body. Proteomic studies of serum with system biology approach are the best ways to detect changes in the pathophysiology in cells [35,36,37]. COPD is a chronic inflammatory illness that shares many of the same signs and symptoms as people who were exposed to SM gas; with the exception that ML is not a progressive condition. Based on analysis of demographic data, the independence and randomization of patients as well as control patients were shown. All patients had notable air trapped in their lungs which is the principal symptom of SM-induced damage known as ML. In the ML, the majority of these significantly altered proteins, according to GO analysis, were involved in inflammatory processes and immune system-associated proteins. Cell adhesion and integrity, as well as cell interaction with the extracellular matrix, and present in pathways, which represent a major roadblock in tissue regeneration process in ML. There were some proteomic studies on SM exposure serum or plasma, but none have used high via the techniques. Haptoglobin isoforms and Amyloid A1 were previously identified as two of the most commonly changed proteins in ML individuals' plasma and BAL fluid [35,36,37,38]. Another study found changes in a wide range of polymorph nuclear cells (PMNCs) proteins, including S100 family members, antioxidant proteins, Serpin B1, and Cronin 1A. An imbalance in protease/anti-protease activity was also observed in PMNCs results in an uncontrolled and unjustifiable response to stimuli and signals from wounded organs [11]. In general, the elevation of antioxidant and inflammatory proteins, as well as the downregulation of protective and preventive proteins, suggests a continuing process of inflammation and remodeling in ML patients. However, several COPD studies have found plasma proteins differently expressed than the control [39,40,41,42]. A new element of continuing pathophysiology of SM-exposed patients was presented. As expected, most differentially expressed proteins are involved in healing and remodeling processes. However, the discovery of several intercellular proteins, such as Thymosin beta-4, which not only have a structural role in cellular components, but also play a part in extracellular matrix(ECM) regeneration and remodeling in healing tissues, casts doubt on earlier assumptions. Thymosin beta-4 is a regenerative peptide linked to wound healing, angiogenesis, cardiomyocyte migration, repair, anti-fibrotic [43,44,45,46,47]. It can regulate the actin polymerization process and act as an anti-inflammatory in human fluids [48, 49]. Compared to HC, high overexpression levels show that tissue regeneration and cell migration are still present in wide areas of wounded tissues. Upregulation of proteins, such as CFHR3, CRP, IGLV3-25, IGLV3-21, IGKV4-1, MPO, FCN2 and TREML1, indicates an activated immune system and an inflammatory or pro-inflammatory response by tissue cells, as well as other body systems in order to orchestrate a coordinated response against cell injury. All of proteins play a distinct role in the cleansing of tissue by cell death, the development of a basal niche for new cell growth, and attraction of effective cells in healing process, which includes angiogenesis and ECM remodeling. Specific proteins, such as TLN1, FGA, ITGB3, PPIA and TUBA1B would be required to regulate cell adhesion and spread following the establishment of conditions for cells to proliferate. Cells may attach to modified ECM and freshly replaced neighbor cells thanks to these substances' ability to modify and control cell connections and integrity. The majority of the proteins that were downregulated are involved in lipid metabolism and circulation in the human body, including APOCI, APOCIII, and adiponectin. Obesity and metabolic syndrome are both linked to these proteins. Reduced levels of these proteins contribute to higher cholesterol and triglyceride levels in the liver and fatty tissue, which can lead to cardiovascular disease. Adiponectin deficiency results in inadequate mitochondrial energy output [50]. It increases insulin sensitivity by inhibiting gluconeogenesis and promoting fatty acid oxidation [51]. The pathogenesis of ML shows a modified continual healing procedure in which cells strive to proliferate and duplicate normal tissue layers, but the lack of a bed to mount the layers leads to constant cell death and destruction of secreted proteins, as well as tissue layers. The presence of defined proteins in the serum indicates that there is ongoing cell damage and that the cells are unable to heal the damaged organ. As previously stated, the presence of many repairs and balancing components in discovered proteins shows that cells always strive to produce suitable conditions for regenerating previously balanced and normal conditions. The absence or the downregulation of critical signals, such as PLXDC2 and fundamental extracellular matrix proteins such as KRT14, as well as the overexpression of proinflammatory proteins, results in an uncontrollable cycle of tissue layer repair and destruction. Histopathological studies, as well as particular immunofluorescent staining of ECM and cytoskeletal elements of biopsies taken from exposed patients, are highly recommended to understand the process and unravel the intricacy of the phenomena. Considering research ethics difficulties, age, and severe to moderate patient conditions, this strategy was not feasible. At the same time as this study was conducted on serum, the proteome analysis of skin biopsy and eye tear samples of ML and HC subjects was performed with the same method and number of samples [52, 53].
Conclusion
SM is an oxidative chemical which quickly reacts with various cell components.
In repairing the damaged parts of lung, epithelial cells, macrophages, and fibroblasts are activated in the damaged area. After decades of exposure to SM, there was a significant rise in the presence of key proteins which secrete in healing cells. The overexpression of adhesion and structural proteins such ITGB3 and TUBA1B cause the damaged cells to try harder to attach to ECM and move to form new tissues. The downregulation of KRT14, which is found in cell–cell adhesions and is a crucial component for cell migration and healing. In ML patients, it appears that the tissue repair cycles are disrupted. Although this event primes cell regeneration and proliferation, it also coordinates the immune system's pro-inflammatory components to set the stage for optimal tissue repair, resulting in a steady stream of inflammatory indicators.
Availability of data and materials
The data which support this study's findings are available from the corresponding author upon reasonable request.
Abbreviations
- SM:
-
Sulphur Mustard
- ML:
-
Mustard Lung
- TMT:
-
Tandem Mass Tag
- EA:
-
Enrichment analysis
- COPD:
-
Chronic pulmonary obstruction
- BAL:
-
Broncho alveolar lavage
- DAPs:
-
Differentially abundant proteins
- GO:
-
Gene ontology
- ATS:
-
American Thoracic Society
- PFT:
-
Pulmonary function testing
- CO:
-
Carbon monoxide
- FEV1% Pred:
-
Predicted forced expiratory volume in one second
- FVE:
-
Forced Vital Capacity
- RT:
-
Room temperature
- APAF:
-
Australian Proteome Analysis Facility
- PBS:
-
Phosphate buffer saline buffer
- DTT:
-
Dithiothreitol
- IAA:
-
Ioadoacetamid
- ACN:
-
Anhydrous acetonitrile
- PPIs:
-
Protein–protein interactions
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- FC:
-
Fold change
References
Dacre JC, Goldman M. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol Rev. 1996;48(2):289–326.
Khateri S, Ghanei M, Keshavarz S, Soroush M, Haines D. Incidence of lung, eye, and skin lesions as late complications in 34,000 Iranians with wartime exposure to mustard agent. J Occup Environ Med. 2003;45(11):1136–43.
Salamati P, Saghafinia M, Abdollahi M. A review on delayed toxic effects of sulfur mustard in Iranian veterans. DARU J Pharmaceut Sci. 2012;20(1):1–8.
Ghanei M, Harandi AA. Mustard lung: diagnosis and treatment of respiratory disorders in sulfur-mustard injured patients. London: Academic Press; 2016.
Najafi A, Ghanei M, Jamalkandi SA. Airway remodeling: systems biology approach, from bench to bedside. Technol Health Care. 2016;24(6):811–9.
Ebadi A, Moradian T, Mollahadi M, Saeed Y, Refahi AA. Quality of life in Iranian chemical warfare veteran’s. Iran Red Crescent Med J. 2014;16(5):e5323.
Tahmasbpour E, Ghanei M, Qazvini A, Vahedi E, Panahi Y. Gene expression profile of oxidative stress and antioxidant defense in lung tissue of patients exposed to sulfur mustard. Mutat Res/Genetic Toxicol Environ Mutagenesis. 2016;800:12–21.
Emad A, Emad Y. Increased in CD8 T lymphocytes in the BAL fluid of patients with sulfur mustard gas-induced pulmonary fibrosis. Respir Med. 2007;101(4):786–92.
Emad A, Emad Y. CD4/CD8 ratio and cytokine levels of the BAL fluid in patients with bronchiectasis caused by sulfur mustard gas inhalation. J Inflamm. 2007;4(1):1–11.
Tahmasbpour Marzony E, Ghanei M, Panahi Y. Oxidative stress and altered expression of peroxiredoxin genes family (PRDXS) and sulfiredoxin-1 (SRXN1) in human lung tissue following exposure to sulfur mustard. Exp Lung Res. 2016;42(4):217–26.
Shahriary A, Mehrani H, Ghanei M, Parvin S. Comparative proteome analysis of peripheral neutrophils from sulfur mustard-exposed and COPD patients. J Immunotoxicol. 2015;12(2):132–9.
Imani S, Salimian J, Bozorgmehr M, Vahedi E, Ghazvini A, Ghanei M, et al. Assessment of Treg/Th17 axis role in immunopathogenesis of chronic injuries of mustard lung disease. J Recept Signal Transd. 2016;36(5):531–41.
Li H, Liu Q, Jiang Y, Zhang Y, Xiao W, Zhang Y. Disruption of th17/treg balance in the sputum of patients with chronic obstructive pulmonary disease. Am J Med Sci. 2015;349(5):392–7.
Xu J-W, Li Y-L, Zhang S-J, Yang W-Q, Nie W-T, Jiang H-Q. Quantitative serum proteomic analysis of essential hypertension using itraq technique. BioMed Res Int. 2017;2017.
Arzoumanian L. What is hemolysis, what are the causes, what are the effects. BD Tech Talk. 2003;2:1–3.
Timms JF, Arslan-Low E, Gentry-Maharaj A, Luo Z, T’Jampens D, Podust VN, et al. Preanalytic influence of sample handling on SELDI-TOF serum protein profiles. Clin Chem. 2007;53(4):645–56.
Richards AL, Eckhardt M, Krogan NJ. Mass spectrometry-based protein–protein interaction networks for the study of human diseases. Mol Syst Biol. 2021;17(1):e8792.
Chen C, Hou J, Tanner JJ, Cheng J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int J Mol Sci. 2020;21(8):2873.
Grossi F, Rijavec E, Genova C, Barletta G, Biello F, Maggioni C, et al. Serum proteomic test in advanced non-squamous non-small cell lung cancer treated in first line with standard chemotherapy. Br J Cancer. 2017;116(1):36–43.
Johannsen C, Koehler CJ, Thiede B. Comparison of LFQ and IPTL for protein identification and relative quantification. Molecules. 2021;26(17):5330.
Kuleš J, Bilić P, Horvatić A, Kovačević A, Guillemin N, Ljubić BB, et al. Serum proteome profiling in canine chronic valve disease using a TMT-based quantitative proteomics approach. J Proteomics. 2020;223: 103825.
Mostafaei S, Borna H, Emamvirdizadeh A, Arabfard M, Ahmadi A, Salimian J, et al. Causal Path of COPD Progression-Associated Genes in Different Biological Samples. COPD J Chronic Obstruct Pulmonary Dis. 2022;19(1):290–9.
Aronson JK. Biomarkers and surrogate endpoints. Br J Clin Pharmacol. 2005;59(5):491.
Wang DL, Xiao C, Fu G, Wang X, Li L. Identification of potential serum biomarkers for breast cancer using a functional proteomics technology. Biomarker Res. 2017;5(1):1–10.
Sun Y, Liu S, Qiao Z, Shang Z, Xia Z, Niu X, et al. Systematic comparison of exosomal proteomes from human saliva and serum for the detection of lung cancer. Anal Chim Acta. 2017;982:84–95.
Crimmins EM, Shim H, Zhang YS, Kim JK. Differences between men and women in mortality and the health dimensions of the morbidity process. Clin Chem. 2019;65(1):135–45.
Parker K, Horowitz JM, Stepler R. On gender differences, no consensus on nature vs. nurture (2017)
Smyth GK. Limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and Bioconductor. Springer; 2005. p. 397-420.
Nobakht M. Gh BF, Hasani Nourian Y, Arabfard M. Identification of shared gene signatures in different stages of nonalcoholic fatty liver disease using integrated microarray datasets. 2022;22(1):e122362.
Franz M, Rodriguez H, Lopes C, Zuberi K, Montojo J, Bader GD, et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46(W1):W60–4.
Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CYJ, Williamson NA, et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15(15):2597–601.
Fonseka P, Pathan M, Chitti SV, Kang T, Mathivanan S. FunRich enables enrichment analysis of OMICs datasets. J Mol Biol. 2021;433(11): 166747.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic acids Res. 2007;36(11):D480–4.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Mehrani H, Ghanei M, Aslani J, Tabatabaei Z. Plasma proteomic profile of sulfur mustard exposed lung diseases patients using 2-dimensional gel electrophoresis. Clin Proteomics. 2010;8(1):1–11.
Koba T, Takeda Y, Narumi R, Shiromizu T, Nojima Y, Ito M, et al. Proteomics of serum extracellular vesicles identifies a novel COPD biomarker, fibulin-3 from elastic fibres. ERJ Open Res. 2021;7(1):00658.
Serban KA, Pratte KA, Bowler RP. Protein biomarkers for COPD outcomes. Chest. 2021;159(6):2244–53.
Mehrani H, Ghanei M, Aslani J, Golmanesh L. Bronchoalveolar lavage fluid proteomic patterns of sulfur mustard-exposed patients. PROTEOM Clin Appl. 2009;3(10):1191–200.
Verrills NM, Irwin JA, Yan He X, Wood LG, Powell H, Simpson JL, et al. Identification of novel diagnostic biomarkers for asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(12):1633–43.
Bozinovski S, Hutchinson A, Thompson M, MacGregor L, Black J, Giannakis E, et al. Serum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(3):269–78.
Rana GSJB, York TP, Edmiston JS, Zedler BK, Pounds JG, Adkins JN, et al. Proteomic biomarkers in plasma that differentiate rapid and slow decline in lung function in adult cigarette smokers with chronic obstructive pulmonary disease (COPD). Anal Bioanal Chem. 2010;397(5):1809–19.
Merali S, Barrero CA, Bowler RP, Chen DE, Criner G, Braverman A, et al. Analysis of the plasma proteome in COPD: Novel low abundance proteins reflect the severity of lung remodeling. COPD J Chronic Obstruct Pulmonary Dis. 2014;11(2):177–89.
Goldstein AL, Kleinman HK. Advances in the basic and clinical applications of thymosin β4. Expert Opin Biol Ther. 2015;15(sup1):139–45.
Li H, Wang Y, Hu X, Ma B, Zhang H. Thymosin beta 4 attenuates oxidative stress-induced injury of spinal cord-derived neural stem/progenitor cells through the TLR4/MyD88 pathway. Gene. 2019;707:136–42.
Xiong Y, Mahmood A, Meng Y, Zhang Y, Zhang ZG, Morris DC, et al. Neuroprotective and neurorestorative effects of thymosin β4 treatment following experimental traumatic brain injury. Ann N Y Acad Sci. 2012;1270(1):51–8.
Wirsching H-G, Krishnan S, Florea A-M, Frei K, Krayenbühl N, Hasenbach K, et al. Thymosin beta 4 gene silencing decreases stemness and invasiveness in glioblastoma. Brain. 2014;137(2):433–48.
Kim J, Wang S, Hyun J, Choi SS, Cha H, Ock M, et al. Hepatic stellate cells express thymosin Beta 4 in chronically damaged liver. PLoS ONE. 2015;10(3): e0122758.
Young JD, Lawrence AJ, MacLean AG, Leung BP, McInnes IB, Canas B, et al. Thymosin β 4 sulfoxide is an anti-inflammatory agent generated by monocytes in the presence of glucocorticoids. Nat Med. 1999;5(12):1424–7.
Banerjee I, Zhang J, Moore-Morris T, Lange S, Shen T, Dalton ND, et al. Thymosin beta 4 is dispensable for murine cardiac development and function. Circ Res. 2012;110(3):456–64.
Bauche IB, El Mkadem SA, Pottier A-M, Senou M, Many M-C, Rezsohazy R, et al. Overexpression of adiponectin targeted to adipose tissue in transgenic mice: impaired adipocyte differentiation. Endocrinology. 2007;148(4):1539–49.
Fisman EZ, Tenenbaum A. Adiponectin: a manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc Diabetol. 2014;13(1):1–10.
Jamshidi V, Gh NM, Fatemeh B, Parvin S, Bagheri H, Ghanei M, et al. Proteomics analysis of chronic skin injuries caused by mustard gas. BMC Med Genomics. 2022;15(1):1–12.
Parvin S, Shahriary A, Aghamollaei H, Gh B, Bagheri H, Ghanei M, et al. Tear proteomics analysis of patient suffered from delayed mustard gas keratopathy. Proteome Sci. 2022;20(1):1–10.
Acknowledgements
This research was supported by the Baqiyatallah University of Medical Sciences, Tehran, Iran. Aspects of this research were facilitated by access to Macquarie University trading as Australian Proteome Analysis Facility (APAF).
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
AN and MGh conceived of the presented idea. ShP and AGh carried out the experiment. ShP carried out data gathering. MA performed the computations. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
This study involving human participants was based on the ethical standards of institutional and national research committee and with 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the Institutional Review Board and the Ethical Committee of Baqiyatallah University of Medical Sciences (code: IR.BMSU.REC.1395.381). All recruited participants signed the informed consent form.
Human and animal ethics
All of experimental procedures involving human were conducted based on Institutional Review Board and the Ethical Committee of Baqiyatallah University of Medical Sciences.
Consent for publication
Not applicable.
Competing interests
Authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. The list of differentially abundant proteins.
Additional file 2
. The details of the PPI network in the Cytoscape tool.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Parvin, S., Arabfard, M., Ghazvini, A. et al. Comparative proteomic analysis of mustard lung as a complicated disease using systems biology approach. BMC Pulm Med 22, 437 (2022). https://doi.org/10.1186/s12890-022-02240-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-022-02240-3