
Abstract
Background
Microbiomics offers new methods for conducting epidemiological surveys of oral microbiota in large populations. Compared to curette sampling, swab sampling is more convenient and less technically sensitive, making it more suitable for such surveys. To verify the feasibility of using swabs for buccal mucosa sampling in large-scale studies, we collected samples from the buccal mucosa and tooth surfaces of healthy individuals using both swabs and curettes. Microbiomics was employed to analyze and compare microbial abundance and diversity between these two methods.
Methods
Four sites were assessed: the buccal mucosa on both sides and the buccal surfaces of the left and right mandibular first molars. Two sampling methods, swab and curette, were used to collect bacterial communities from healthy individuals. Specifically, buccal mucosa samples (n = 10) and tooth surface samples (n = 20) were analyzed using 16 S rDNA gene sequencing. Bacterial signals were detected through fluorescence in situ hybridization (FISH), targeting the bacterial 16 S rDNA gene. Metastats analysis and Wilcoxon test were used.
Results
A total of 383 OTUs were detected in the 30 samples, which belonged to 1 kingdom (bacteria), 11 phyla, 23 classes, 40 orders, 75 families, 143 genus, and 312 species. Among them, 223 OTUs were found on both the buccal mucosa and tooth surfaces. The statistics suggest that although there were no significant differences in colony composition, there were differences in the abundance and distribution of colonies on the dental and buccal mucosal surfaces. When detecting oral disease-causing pathogens such as Enterococcus faecalis and Porphyromonas gingivalis, the efficiency of detection is higher when using curette sampling. Compared to right tooth sampling with a curette, the swab sampling group had higher levels of Firmicutes, while Fusobacteria and Bacteroidetes were more prevalent in the curette tissues.
Conclusions
In oral health individuals, there is no difference in the bacterial composition of the oral buccal mucosa and the dental surface, differing only in abundance. Thus, the buccal mucosa can act as a substitute for the teeth in epidemiological investigations exploring the bacterial composition of the oral cavity.
Similar content being viewed by others

Background
According to a report by Mark Welch et al. [1], there are over 700 prokaryotic species that inhabit the human oral cavity. The oral microbiome plays a crucial role in maintaining not only oral health, but also overall bodily health [1, 2]. The human body is known to harbor numerous microorganisms that are associated with both health and disease [1, 3]. Several studies have suggested that oral microbiomes may be linked to various systemic diseases [3,4,5].
Research has shown that bacteria reside in specific niches within the human mouth [1, 6]. Moreover, differences in local sites within the oral cavity can lead to distinct distribution patterns as revealed by 16s rRNA sequencing [7,8,9,10]. Current studies are focused on bacterial colonization of various essential areas of the oral cavity, including dental surfaces, buccal mucosa, saliva, and the tongue [1, 2, 6]. However, unifying sampling methods in large-scale studies poses challenges, as there is a lack of standardized sampling protocols and regions for healthy individuals. Therefore, our upcoming research aims to address this gap [11, 12]. Investigating the microbiome that represents oral health in large populations is time-consuming and labor-intensive, highlighting the urgent need for a standardized reference. When it comes to the health of an individual, major bacterial sequencing is identical, supporting the opinion that a core microbiome exists in healthy individuals [13, 14].
The Human Microbiome Project Consortium revealed that the unique feature of an individual’s microbiome is stable and characteristic of healthy people [7, 15]. Accumulating evidence has shown that bacteria can choose the habitat that suit them best [1, 6, 13, 16]. At the micron scale, the distribution between Gram-positive and Gram-negative bacteria differs [17]. A study based on five individuals proved that Streptococcus mitis and Gemella hemolysans are the predominant species of the buccal epithelium [18]. Meanwhile, different niches are meaningful for studying the representatives of the oral microbiome [1, 19]. Selecting different sampling areas and techniques in the oral cavity is crucial for research and clinical applications. It was once believed that saliva represents the microbiome on the oral surface. However, Segata and his colleagues’ study implied that saliva has more bias on the tongue and palate [20]. The oral cavity’s diverse microenvironments, such as teeth and buccal mucosa, each host distinct microbial compositions. Using swabs or curette sampling techniques appropriately ensures comprehensive and reliable data, as recommended by the World Health Organization [21]. Efficient sampling is vital for epidemiological surveys, especially under time constraints. Optimized sampling strategies reduce survey time and resources, crucial for timely data collection during outbreaks or monitoring infectious diseases. Swab sampling is more convenient and faster than curette sampling, making it ideal for large-scale surveys.
Therefore, our hypothesis is that in individuals with good oral health, swab sampling will show no significant variation in the distribution of oral core microbiota across different regions of the oral cavity. This explores the feasibility of using swab sampling instead of curette sampling in large populations of healthy individuals.
Methods
Study design and participants
Five persons with no oral diseases were enrolled at the Stomatology Hospital of Tianjin Medical University, and informed consent was obtained from all subjects. The inclusion criteria is as follows: (1) People who had no oral diseases particularly caries, periodontitis, and oral mucosal diseases. (2) Teeth without previous restorations. Exclusion criteria is as follows: (1) People who are pregnant. (2) People who suffer from severe systematic diseases, such as diabetes, immune system diseases, and genetic diseases. (3) People who have taken antibiotics within two weeks before sampling.(4) People who have had or currently receiving radiotherapy or chemotherapy. (5) People with complete or incomplete absence of dentition. (6) Individuals who smoke or have smoked within the past six months.
Sample collection and clinical grouping
Ultimately, five teeth were included. One trained clinician examined the cases and collected the samples under aseptic conditions. We prevented subjects from eating and drinking for at least 2 h before sampling. Two sampling methods successively sampled each patient. The buccal plaque of the left and right lateral mandibular first molars was taken with curettes and swabs, respectively, and the buccal mucosa of the left and right lateral teeth was taken with swabs, respectively. Finally, the colonies of the left buccal swab, left tooth curette, left tooth swab, right buccal swab, right tooth curette, and right tooth swab were counted. All samples were transported to the laboratory on ice within 2 h of collection. Moreover, they were stored at -80℃ in a refrigerator before 16 S rDNA sequencing.
16 S rDNA sequencing
PCR (TransGenAP221-02: Transstart Fastpfu DNA Polymerase) was used to amplify the extracted genomic DNA.DNA concentration and purification were monitored on 1% agarose gels. The whole gene sequencing was conducted on the Miseq platform. Synthesize specific primers with barcodes according to the designated sequencing region, or synthesize fusion primers with mispaired bases. In our study, we used universal primers targeting the 16S rRNA gene for bacterial identification and diversity analysis. Specifically, the primers used were: Forward Primer: 338F(5’-ACTCCTACGGGAGGCAGCAG-3’); Reverse Primer: 806R(5’-GGACTACHVGGGTWTCTAAT-3’). These primers are well-established in microbiome research for their broad range and ability to amplify bacterial DNA across a wide variety of species. This choice was made to ensure a comprehensive analysis of the microbial communities present in the oral cavity.
Primers performed PCR with barcodes, which targeted the V3-V4 region of the 16 S rDNA region. PCR was performed under the following conditions: 94℃ for 5 min, followed by 28 cycles of 94℃ for 30 s, 55℃ for 30 s, and 72℃ for 1 min, and then a final elongation step at 72℃ for 7 min and 4℃ in the end. Using fast length adjustment of short reads (FLASH) software merged primitive DNA sequence. Using the database SRA (Sequence Read Archive, http://www.ncbi.nlm.nih.gov/Traces/sra) established by the NCBI data center to store shared raw sequencing data. The Quantitative Insights Into Microbial Ecology (QIIME) software were applied to analyze the results of sequencing. The data obtained from the MiSeq sequencing is in the form of paired-end sequences.
Data management and statistical analysis
To enhance data processing efficiency, we assigned numbers to samples collected in the same manner. The left tooth curette sample was labeled as AZ, the left buccal swab as AS, the left tooth swab as AX, the right tooth curette as BS, the right buccal swab as BZ, and the right tooth swab as BX. AZ comprised of samples A1, A4, A7, A11, and A13. AS comprised of samples A2, A5, A8, A10, and A14. AX comprised of samples A3, A6, A9, A12, and A15. BS comprised of samples A17, A20, A23, A25, and A29. BZ comprised of samples A16, A19, A22, A26, and A28. BX comprised of samples A18, A21, A24, A27, and A30 (Table S1).
Alpha diversity was used to estimate the richness and diversity of microbial species. We use QIIME (Version 1.8), Uclust (Version 1.2.22), and Usearch (Version 10.0.240) to analyze the results and generate rarefaction curves, and the R package to visualize the difference of each sample. Beta diversity was analyzed to evaluate differences among samples in species complexity. We used Qiime to analyze the results and rarefaction curves, and the R package to exhibit the differences of each sample. Weighted UniFrac distance was calculated using Qiime. Anonymous analysis was used to compare differences among groups with differences within groups. Linear discriminant (LDA) effect size analysis was performed, setting the LDA score to 3 by default (Fig S1). P-values less than 0.05 were considered statistically significant.
Results
The overall diversity of bacterial communities across samples
Initially, the obtained FASTQ data is filtered by removing the bases with quality scores below 20 from the end of the reads. A 50 bp window is set, and if the average quality score within the window is below 20, the bases at the end of the window are trimmed. Reads with less than 50 bp after quality control are filtered out. Then, the paired sequences are merged into a single sequence based on their PE sequencing overlap relationship. The raw data (Raw PE) is processed by removing the barcodes and primers and merging them to obtain raw tags. The specific result statistics are shown in Table S2. In our study, we collected 30 specimens for each of the 5 individuals who had a healthy oral status. The baseline information of the subjects is presented in Table 1, while the distribution statistics of high-quality sequences obtained after data preprocessing are shown in Table S3.
We utilized various indexes, namely Shannon, Specaccum, and Chao1, to estimate the α-diversity. The rarefaction curves of observed OTUs per sample indicated that additional sequencing would be necessary to capture all phylotypes (Fig. 1A). The Shannon-Wiener curve had reached a plateau, suggesting that the sample size was sufficient to capture species diversity (Fig. 1B). The species accumulation curves reflected the biodiversity within the sample. In this experiment, the total number of OTUs detected increased steadily with the increase of the sample size, and the trend of increase was flat. This indicated that the sample size in this experiment was sufficient to support the conclusion. The results showed that regardless of the sampling tools used, the microbiome diversity between the tooth surface and buccal mucosa was different. The group that took the right buccal mucosal swab sample had a faster decrease in the Rank-Abundance curve than the other groups, indicating that collecting samples in this way in clinic may result in uneven bacterial diversity. The most abundant sample content was obtained when the left side teeth were scraped and sampled. As revealed by the Shannon diversity index, there were significant differences in diversity between BZ and AZ or BS. The AZ-BS, AS-BZ, and Shannon values were statistically different from the AS-BZ group (P = 0.004, P = 0.003; Fig. 1C). This indicates that the biodiversity exhibited by the right buccal swab sampling was not abundant, and the biodiversity between the buccal mucosa and tooth surface was uneven. Overall, the sampling results of the AZ group were significantly better than the other groups in terms of microbial diversity and the authenticity of the measured data. The OTUs detected by collecting bacteria on teeth were higher than those collected on the buccal mucosa.

Description of oral microbiota structures in the samples. (a) Rarefaction analysis of bacterial 16 S rDNA gene sequences was applied. Suggested by a plateau, it told us whether additional taxa were found. Different colors represent different samples. (b) Shannon index curves were performed to assess whether the samples were sufficient, indicated by a plateau. (c) Box plots depict differences in bacterial diversity among AZ-AX, AS-BZ, BS-BX. BS represents the right tooth curette; AZ represents the left tooth curette; BZ represents the right buccal swab; AS represents the left buccal swab; AX represents the left tooth swab; BX represents the right tooth swab
Taxonomic analysis in each group
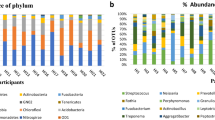
Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, and Fusobacteria were the top five most abundant phyla, accounting for 98.26% of all sequences. Firmicutes was the most abundant phylum, comprising 41.25% of sequences (Fig. 2A). Neisseria, Streptococcus, Haemophilus, Veillonella, and Rothia were the five most abundant genera at the genus level (Fig. 2B).

Composition of bacterial communities across samples at the phylum and genus levels. (a) Relative abundance of bacterial phyla among each group. (b) Relative abundance of bacterial genus among each group. BS represents the right tooth curette; AZ represents the left tooth curette; BZ represents the right buccal swab; AS represents the left buccal swab; AX represents the left tooth swab; BX represents the right tooth swab
Association between groups of samples and indication of specific pathogens detection
Compared with right tooth sampling with a curette, swab sampling is more efficient in detecting Fusobacteria (Fig. 3A). There was a difference in abundance, including Bacteroidetes, Firmicutes, and Fusobacteria between AS-BZ/AZ-BS (Fig. 3B). Bacteroidetes were more abundant in the left tooth compared to the right. Heatmap analysis was used to exhibit the pathogen that was considered to lead to the disease. As the outcome revealed, Bacteroidetes, Fusobacteria, Spirochaetes, Cyanobacteria, Firmicutes, and Actinobacteria were part of the top 20 phyla (Fig. 4A). The Firmicutes and Proteobacteria were measured to have a similar distribution in each group. Prevotella intermedia was more abundant in the AX and AZ groups. Enterococcus faecalis and Porphyromonas endodontalis, two pathogens known to cause pulp diseases, showed significant abundance in the samples taken from the tooth surface (Fig. 4B). There was little difference in the sampling method for testing these two bacteria alone.

The taxonomic analysis of microbiome relations. (a) The Wilcoxon test of AX-BX and AS-BZ of the group on the phylum level. (b) The Wilcoxon test of AX-BX and AZ-BS of the group on the phylum level. BS represents the right tooth curette; AZ represents the left tooth curette; BZ represents the right buccal swab; AS represents the left buccal swab; AX represents the left tooth swab; BX represents the right tooth swab

The taxonomic analysis of colony composition. (a) The heatmap of the top 20 species of each group. (b) The heatmap of bacterial genera. BS represents the right tooth curette; AZ represents the left tooth curette; BZ represents the right buccal swab; AS represents the left buccal swab; AX represents the left tooth swab; BX represents the right tooth swab
Metastats analysis for inter-group significance indicated that the differences between groups were greater than the differences within groups, suggesting that the grouping was meaningful (Table 2). Buccal mucosa and tooth surface microorganisms share a great degree of similarity in composition and structure (Fig. 5A and B). The colonies obtained by the two methods of sampling at the teeth were more similar (Fig. 5C).

Interpretation of Intra-Group Taxa. (a) Unifrac distance analysis of each group. (b) NMDS of oral microbiota in groups. (c) PCA analysis in different groups. BS: right tooth curette; AZ: left tooth curette; BZ: right buccal swab; AS: left buccal swab; AX: left tooth swab; BX: right tooth swab
After the LDA test, no significant difference was detected between the AX and BX groups (Fig. 6A). LDA analysis showed a significant difference in the Mycobacteria among the AS, AZ, BS, and BZ groups. In the AS group, Mycobacteriaceae had a high LDA score. In the samples taken from AZ, there were significant differences in the detection of Cardiobacterium, Corynebacterium, and Capnocytophaga. Among the samples obtained from the BS group, Fusobacteria and Porphyromonadaceae had a high LDA score. In the BZ group, the significant difference between Lactobacillus and Streptococcus was higher than in other groups. In the BS-BX group, the class level of Betaproteobacteria and Fusobacteria, the genus level of Neisseria, Pseudopropionibacterium, Leptotrichia, Corynebacterium, and Porphyromonas, the order level of Neisseriales, Fusobacteria, and Corynebacteriales, the family level of Neisseriaceae, Leptotrichiaceae, Corynebacteriaceae, and Porphyromonadaceae, and the species level of Pseudopropionibacterium propionicum, Corynebacterium matruchotii, Neisseria elongata, and Leptotrichia 225 were more abundant than in the tooth group (Fig. 6B). As for swab-sampling on teeth, the genus level of Capnocytophaga had a high LDA score. As for buccal mucosa, the species level of Mycobacterium neoaurum, Haemophilus 036, and Streptococcus 071, the family level of Streptococcaceae, the genus level of Haemophilus and Streptococcus, the order level of Lactobacillales, the phylum of Firmicutes, and the class level of Bacilli had a high LDA score. Bacteroidetes and Fusobacteria on the phylum level, and Porphyromonadaceae on the family level, were more abundant. The LDA score is high for Firmicutes on the phylum level, Bacilli on the class level, and Streptococci on the genus level. (Fig. 6C)

LDA Analysis of the Samples. (a) LDA scores predict gene function enriched in different groups. BS: right tooth curette; AZ: left tooth curette; BZ: right buccal swab; AS: left buccal swab; AX: left tooth swab; BX: right tooth swab. (b) Analysis of each group. (c) Difference between AS-BZ and AZ-BS groups. BS: right tooth curette; AZ: left tooth curette; BZ: right buccal swab; AS: left buccal swab; AX: left tooth swab; BX: right tooth swab
Discussion
The study aimed to analyze the composition and distribution of microbial communities in the buccal mucosa and tooth surfaces within the healthy oral cavity. The findings from this investigation provide valuable insights into the microbial diversity and its implications for oral health research.
The results indicated that there is a considerable similarity in the microbial composition between the buccal mucosa and tooth surfaces, with a total of 223 OTUs found in both regions. This suggests that these two sites share a core microbiome, despite variations in microbial abundance. Firmicutes were more prevalent in swab samples, while Fusobacteria and Bacteroidetes were more abundant in curette samples from the tooth surface. This distribution aligns with previous studies highlighting the dominance of these phyla in different oral niches [1, 22, 23].
The presence of significant microbial taxa such as Prevotella intermedia and Porphyromonas endodontalis on the tooth surface indicates the potential for using specific sampling methods to detect oral pathogens effectively. This is crucial for diagnosing and monitoring oral diseases. The study’s detection of these pathogens is consistent with findings from other research that identifies specific niches within the oral cavity for different bacterial species [2, 7, 18, 24].
Swab samples were found to be easier and less invasive to collect compared to curette samples, making them more suitable for large-scale epidemiological studies. However, the detection of disease-associated pathogens such as Enterococcus faecalis and Porphyromonas gingivalis was more efficient with curette sampling. This is consistent with previous studies, which suggest that the method of sampling can significantly impact the detection and analysis of microbial communities [4].
The similarities in microbial composition between the buccal mucosa and tooth surfaces support the use of buccal mucosa sampling as a representative method for studying the oral microbiome. This approach is particularly advantageous for large-scale studies due to its non-invasive nature and ease of collection. However, the differences in microbial abundance and the efficiency of pathogen detection between the two sampling methods highlight the need for careful consideration of the sampling technique based on the research objective. For instance, while swab samples are ideal for general microbiome studies, curette samples may be preferred for pathogen-specific investigations.
Research by the Human Microbiome Project Consortium has revealed that the unique feature of an individual’s microbiome is stable and characteristic of healthy people, supporting the idea of a core microbiome in healthy individuals [3, 21]. This consistency further validates the use of buccal mucosa sampling as a reliable method for microbiome studies.
While this study provides a comprehensive comparison of microbial communities in different oral regions, it has several limitations. The sample size was relatively small, which may affect the generalizability of the findings. Future studies should include a larger and more diverse population to validate these results. Additionally, incorporating other oral sites such as the tongue and saliva could provide a more complete understanding of the oral microbiome.
In conclusion, our study underscores the importance of selecting appropriate sampling sites and methods to accurately characterize the oral microbiome. These findings contribute to the foundation for future research and clinical applications aimed at understanding the role of the oral microbiome in health and disease. Effective sampling strategies are essential for obtaining reliable data, particularly in large-scale epidemiological surveys where time and resources are limited. Swab sampling’s convenience and efficiency make it an ideal choice for such studies, although curette sampling may be necessary for more detailed pathogen analysis.
Conclusions
Our study provides preliminary evidence that there is no significant difference between oral buccal mucosa and hard surfaces for bacteria composition. This may be used in oral epidemiological surveys of large healthy populations. The detection of the distribution of pathogenic bacteria in oral cavity must utilize the appropriate sampling method depending on the bacteria under study.
Data availability
The data that support the findings of this study are available on request from the corresponding author, Dy Liu. The datasets generated and analyzed during the current study are available in the Sequence Read Archive (SRA) database established by NCBI (https://dataview.ncbi.nlm.nih.gov/object/PRJNA935080?reviewer=itsp0avuq1bsla1gn5ksurmkc2) and will be made publicly available upon publication. (BioProject ID: PRJNA935080)
Abbreviations
- BS:
-
Represents the right tooth curette
- AZ:
-
Represents the left tooth curette
- BZ:
-
Represents the right buccal swab
- AS:
-
Represents the left buccal swab
- AX:
-
Represents the left tooth swab
- BX:
-
Represents the right tooth swab
References
Mark Welch JL, Ramirez-Puebla ST, Borisy GG. Oral microbiome geography: micron-scale habitat and niche. Cell Host Microbe. 2020;28:160–8.
Verma D, Garg PK, Dubey AK. Insights into the human oral microbiome. Arch Microbiol. 2018;200:525–40.
Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell. 2018;9:488–500.
Zhao H, Chu M, Huang Z, Yang X, Ran S, Hu B, et al. Variations in oral microbiota associated with oral cancer. Sci Rep. 2017;7:11773.
Matsha TE, Prince Y, Davids S, Chikte U, Erasmus RT, Kengne AP, et al. Oral microbiome signatures in diabetes mellitus and periodontal disease. J Dent Res. 2020;99:658–65.
Simon-Soro A, Tomas I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. Microbial geography of the oral cavity. J Dent Res. 2013;92:616–21.
Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–17.
Moon JH, Lee JH. Probing the diversity of healthy oral microbiome with bioinformatics approaches. BMB Rep. 2016;49:662–70.
Zhang SM, Tian F, Huang QF, Zhao YF, Guo XK, Zhang FQ. Bacterial diversity of subgingival plaque in 6 healthy Chinese individuals. Exp Ther Med. 2011;2:1023–29.
Willis JR, Gabaldon T. The human oral microbiome in health and disease: from sequences to ecosystems. Microorganisms. 2020;8:1–29.
Okada A, Sogabe K, Takeuchi H, Okamoto M, Nomura Y, Hanada N. Characterization of specimens obtained by different sampling methods for evaluation of periodontal bacteria. J Oral Sci. 2017;59:491–98.
Matic Petrovic S, Cimbaljevic M, Radunovic M, Kuzmanovic Pficer J, Jotic A, Pucar A. Detection and sampling methods for isolation of Candida spp. from oral cavities in diabetics and non-diabetics. Braz Oral Res. 2015;29:1–7.
Xu X, He J, Xue J, Wang Y, Li K, Zhang K, et al. Oral cavity contains distinct niches with dynamic microbial communities. Environ Microbiol. 2015;17:699–710.
Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy core microbiome of oral microbial communities. BMC Microbiol. 2009;9:259.
Human Microbiome Project. A framework for human microbiome research. Nature. 2012;486:215–21.
Elizabeth KC, Christian LL, Micah H, Noah F, Jeffrey IG, Rob K. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–97.
Eren AM, Borisy GG, Huse SM, Mark Welch JL. Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci USA. 2014;111:E2875–84.
Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–32.
Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16:745–59.
Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012;13:42.
The Human Microbiome Project Consortium. NIH Human Microbiome Project. In: NIH Human Microbiome Project. 2010. https://www.hmpdacc.org/doc/HMP_Clinical_Protocol.pdf. National Institutes of Health (NIH). Accessed 10 February 2021.
Keijser BJ, van den Broek TJ, Slot DE, van Twillert L, Kool J, Thabuis C, et al. The impact of maltitol-sweetened chewing gum on the dental plaque biofilm microbiota composition. Front Microbiol. 2018;9:381.
Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, et al. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008;87:1016–20.
Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010;4:962–74.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 81673166, 81372118, 81372467, 81302422 to Kaijun Niu and 82071079 to Dayong Liu), the Key Technologies R&D Program of Tianjin (Key Project: No. 11ZCGYSY05700, 12ZCZDSY20400, 13ZCZDSY20200, and 15YFYZSY00020), and the Science and Technology Project of Tianjin Municipal Health Commission (ZC20132,ZC20134, and ZC0178).
Author information
Authors and Affiliations
Contributions
Y.C. Liu contributed to literature search, conception, design of the study, and drafting of the manuscript. F. Qiao, G. Meng, Y.Q. Gu, Z.Y. Wang and H.M. Wu conducted the literature review and helped to prepare the Introduction and Methods sections of the text. F. Qiao, D.Y. Liu, and K.J. Niu designed the analytical strategy and helped to interpret the findings. D.Y. Liu and K.J. Niu contributed to conception, design, and interpretation, and critically revised the manuscript. All authors gave their final approval and agreed to be accountable for all aspects of the work. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The ethics committee of the Stomatology Hospital of Tianjin Medical University approved this study (ID: TMUhMEC20221105). Five persons with no oral diseases were enrolled at the Stomatology Hospital of Tianjin Medical University, and informed consent was obtained from all subjects. The study was obtained from the administration of Stomatology Hospital of Tianjin Medical University. Written informed consent was provided by all participants. The study’s purpose was clearly explained, and participation was entirely voluntary.
Consent for publication
Not Applicable.
Conflict of interest
No potential conflict of interest was reported by the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.


Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Liu, Y., Qiao, F., Wang, Z. et al. Analysis of the microbial community diversity in various regions of the healthy oral cavity. BMC Oral Health 24, 978 (2024). https://doi.org/10.1186/s12903-024-04677-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-024-04677-w