
Abstract
Background
Habitat transitions have considerable consequences in organism homeostasis, as they require the adjustment of several concurrent physiological compartments to maintain stability and adapt to a changing environment. Within the range of molecules with a crucial role in the regulation of different physiological processes, neuropeptides are key agents. Here, we examined the coding status of several neuropeptides and their receptors with pleiotropic activity in Cetacea.
Results
Analysis of 202 mammalian genomes, including 41 species of Cetacea, exposed an intricate mutational landscape compatible with gene sequence modification and loss. Specifically for Cetacea, in the 12 genes analysed we have determined patterns of loss ranging from species-specific disruptive mutations (e.g. neuropeptide FF-amide peptide precursor; NPFF) to complete erosion of the gene across the cetacean stem lineage (e.g. somatostatin receptor 4; SSTR4).
Conclusions
Impairment of some of these neuromodulators may have contributed to the unique energetic metabolism, circadian rhythmicity and diving response displayed by this group of iconic mammals.
Similar content being viewed by others


Background
Evolutionary transitions, understood as the acquisition of a novel lifestyle within a lineage (e.g. [1]), encompass drastic modifications in several physiological systems (e.g. [2, 3]), to sustain the impacts of a novel environment in physiological homeostasis. Cetacea, an iconic group of aquatic mammals composed of baleen whales (Mysticeti), toothed whales, dolphins and porpoises (Odontoceti), are a “poster child for macroevolution” and typify such an evolutionary process [4]. The number of phenotypic modifications and adaptive traits associated with this land-to-water evolutionary transition is astonishing and implied a profound reorganization in different organ systems (e.g. [4,5,6]; Fig. 1). In fact, many cetacean physio-anatomical traits set them apart as unique among mammals. One remarkable example is their circadian rhythmicity, which was adapted by abolishing neurochemical signals like the hormone melatonin and the neuropeptide cortistatin [5, 7,8,9]. This alteration was accompanied by anatomical and physiological rearrangements, as evidenced by their lower cortical orexinergic bouton density, when compared to closely related terrestrial artiodactyls [10]. Yet, the precise molecular events underpinning the coalescence of these interconnected systems into novel physiological states remain largely unexplored. Among the various chemical messengers modulating physiological functions, neuropeptides are central in regulating various physiological mechanisms [11]. They are defined as short sequences of amino acids synthesized and released by neurons or glia. Moreover, they are responsible for slow-onset, long-lasting modulation of synaptic transmission, acting on neighbouring cells via G protein-coupled receptors (GPCRs) [12].

Evolutionary transitions and pleiotropic molecules. Land-to-water habitat transition experienced by marine mammals such as Cetacea was accompanied by remarkable anatomical, physiological and behavioural adaptations. In this study, we aim to assess the coding status of several neuropeptides with distinct physiological roles. Fossil timeline (grey bars) depicts the estimated ages of the appearance and extinction of key cetacean ancestors. Dating of these events was obtained in the following references [100,101,102,103,104,105,106,107]. In the lower panel, references for each reported role associated with neuropeptides are provided by [14, 18, 26, 47, 53, 56, 58, 60, 66, 108,109,110,111,112,113,114,115,116,117,118]
In this study, we delve into the evolution of several neuropeptide and receptors, with an important role in the coordination of different physiological compartments that have been clearly modified in Cetacea: including the liver, lungs, heart and brain (Fig. 1; [13]). It is worth noting that neuropeptides and their receptors often exhibit pleiotropic effects, implying a potential wide range of physiological functions. As such, neuropeptide FF-amide peptide precursor (NPFF) seems a major player in pain modulation [14, 15], but also has a role in cardiovascular regulation (e.g. [16]). Pyroglutamylated RFamide peptide (QRFP) and its receptor (QRFPR) play a role in the control of feeding behaviour and also participate in sleep regulation [17,18,19]. Neuropeptide B (NPB) and one of its receptors (NPBWR2) are also important for the control of feeding behaviour, as well as pain modulation and sleep regulation [20,21,22,23]. Neuropeptide S (NPS) and its receptor (NPSR1) are central to sleep regulation and are linked to the control of feeding behaviour [24,25,26]. Neuromedin B receptor (NMBR) has been linked with both hormonal regulation and immunity control [27, 28]. Somatostatin receptor 4 (SSTR4) plays a role in hormonal regulation and in stress responses [29]. Finally, neuropeptide Y6 receptor (Npy6r) was associated with hormonal regulation and control of feeding behaviour [30]. Such pleiotropy raises the critical question on how these regulators evolved in such a drastic ecological transition as that experienced in Cetacea evolution. Strikingly, by expanding the analysis to non-Cetacea 161 mammalian species (22 taxonomic orders; Additional file 1: Table S1), we also revealed a complex mutational landscape, possibly indicative of distinct adaptations to multiple environments.
Results
Variable patterns of sequence alterations are found in two RFamide neuropeptides and their receptors
Comparative analysis of the neuropeptide FF-amide peptide precursor (NPFF) exonic sequences revealed localized events of gene pseudogenization across the mammalian tree. More specifically, 38 mammal species presented a PseudoIndex higher than 2 (Additional file 1: Table S2). This built-in assistant metric incorporated in PseudoChecker [31] assesses the sequence erosion status of the tested genes on a discrete scale ranging from 0 (coding) to 5 (pseudogenized).
Subsequent manual annotation allowed the identification of non-conserved open reading frame (ORF) disruptive mutations in both Mysticeti and Odontoceti species, including loss of start codon or indels in exons 1 and 2 (Additional file 1: Table S3). When expanding our search to other mammalian lineages, deleterious mutations were found in kangaroo rats (Dipodomys spp.), Hawaiian monk seal (Neomonachus schauinslandi) and flying foxes (Pteropus spp.) (Fig. 3); in the latter a conserved mutation was validated by SRA searches (Additional file 2: Figs. S1–S4). Also, although no inactivating mutations were discovered, exons 1 and 2 in Canidae (Canis spp. and Vulpes spp.) were not found (Additional file 1: Table S3).
Next, we sought to characterize the coding status of both NPFF receptors. PseudoChecker analyses for neuropeptide FF receptor 1 (NPFFR1) resulted in 127 species presenting a PseudoIndex > 2, contrasting with neuropeptide FF receptor 2 (NPFFR2) where only 48 species seem to yield eroded genes (Additional file 1: Tables S4 and S5). Annotation of collected cetacean genomic sequences revealed extension of the NPFFR1 gene across all analysed species, due to a conserved 11 nucleotide deletion in exon 4 (Fig. 2; Additional file 1: Table S6). For NPFFR2, most of the cetaceans displayed several disrupting mutations including a conserved 2 nucleotide deletion in exon 3 within Delphinoidea (oceanic dolphins, porpoises, beluga and narwhal) and within Physeteroidea (sperm whales) (Fig. 2; Additional file 1: Table S7). In both these receptors, the majority of the ORF-disrupting mutations are found in the last exon, which translates in ~ 20% of the total gene content completely modified (for NPFFR1) and for NPFFR2 more than 25% of the total gene length is not transcribed given the change of frame due to a frameshift mutation.

Pseudogene annotation in Cetacea. Examples of ORF-disrupting mutations in Cetacea for the study genes and respective sequence alignment. Each box represents an exon and lines represent intronic regions. An exhaustive description of the reported disruptive mutations can be seen in Additional file 1: Tables S6, S7, S9, S11, S13, S15, S17, S19 and S21
In other mammalian species, a highly eroded NPFFR1 was also found within Chiroptera (bats), where the majority of the analysed species exhibited several conserved ORF-abolishing mutations and/or lack of exons (Fig. 3; Additional file 1: Table S6). Moreover, ORF-disrupting mutations were also identified in other mammals; however, data from independent SRAs only validated mutations in Canis spp., red fox (Vulpes vulpes) and naked mole-rat (Heterocephalus glaber) (Additional file 1: Table S7). SRA validations for NPFFR1 and NPFFR2 are presented in Additional file 3 (Figs. S1–S10) and Additional file 4 (Figs. S1–S11).

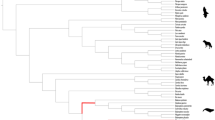
Schematic representation of gene loss events along the mammalian tree. Phylogenetic relationships were adapted from Vazquez [119]. For each gene, we represented in green the species where no ORF-disrupting mutations (frameshift mutations, in-frame premature stop codons, loss of canonical splicing site) were found. On the other hand, species presenting ORF-disrupting mutations are highlighted in red. In orange are symbolized cases where there is some mutational evidence, however the coding status cannot be entirely deduced (e.g. inactivating mutations that occur at the beginning or end of a gene). True absence of exons in the study genes is illustrated in blue. Species presenting (at least) one exon with poor alignment identity with the corresponding reference orthologue are represented in grey. Finally, question marks represent the cases where the absence of a given exon either likely results from the fragmentation (Ns) or incompleteness of the scaffold in the genomic region containing the target gene. An exhaustive description of the reported disruptive mutations can be seen in Additional file 1: Tables S3, S6, S7, S9, S11, S13, S15, S17, S19, S21, S23 and S25
Within Cetacea, PseudoChecker analyses for pyroglutamylated RFamide peptide (QRFP) resulted in 20 species presenting a PseudoIndex higher than 2 (Additional file 1: Table S8). In cetaceans, most of the species showed either an eroded or sequence-modified QRFP with conserved mutations within Mesoplodon spp. (beaked whales) and Delphinidae (Fig. 2; Additional file 1: Table S9). Specifically for baleen whales and oceanic dolphins presenting only mutations at the beginning of the gene, we may not rule out the possibility of having a functional QRPF in these species, as the peptide does not suffer any change of frame while transcribed. Investigation of QRFP inactivation in other mammalian lineages revealed episodes of gene loss (Fig. 3). In particular, QRFP was found to be inactivated and subsequently validated in species such as Pteropus spp.—with the presence of a conserved 1 nucleotide deletion among members of this group—lemurs (Lemuroidea) and echidna (Tachyglossus aculeatus) (Additional file 1: Table S9 and Additional file 5: Figs. S1–S15). In the case of pyroglutamylated RFamide peptide receptor (QRFPR), 64 species presented signs of loss of the in-study gene (Additional file 1: Table S10), comprising mostly cetaceans and chiropterans (Figs. 2 and 3). Accordingly, in all analysed cetaceans the orthologous exon 2 of QRFPR was not found and the exon 5 was either not found or exhibited a conserved in-frame premature stop codon (Additional file 1: Table S11). Moreover, flying foxes showed, once more, evidence of inactivation with several conserved mutations in their coding reading frame (Additional file 1: Table S11). Other examples of species presenting a sequence-altered QRFPR include the California sea lion (Zalophus californianus) and echidna (Fig. 3; Additional file 1: Table S11 and Additional file 6: Figs. S1–S12).
Convergent gene loss among marine mammals of the neuropeptide S and receptor
We next examined the coding status of neuropeptide S (NPS) and neuropeptide S receptor 1 (NPSR1) in mammals to investigate independent gene loss events across the mammalian phylogeny. Although 96 species presented a PseudoIndex higher than 2 for NPS (Additional file 1: Table S12), only odontocetes (toothed cetaceans) except sperm whale (Physeter macrocephalus) and Indus River dolphin (Platanista minor), and West Indian manatee (Trichechus manatus latirostris) exhibited valid truncating mutations (Fig. 3; Additional file 1: Table S13 and Additional file 7: Figs. S1–S5). Regarding NPSR1, PseudoChecker analyses reported a total of 85 species labelled with a PseudoIndex higher than 2 (Additional file 1: Table S14). Annotation of collected genomic sequences revealed NPSR1 gene erosion mainly in two groups: in toothed cetaceans and aquatic carnivores (pinnipeds and otters) (Figs. 2 and 3). While in the first group pseudogenization was confirmed either by the absence of several exons and/or the presence of conserved deleterious mutations, for the latter different ORF-disrupting mutations were retrieved across species (Additional file 1: Table S15). In addition to these two groups, we also found evidence of NPSR1 inactivation in Spanish mole (Talpa occidentalis), Damaraland mole-rat (Fukomys damarensis) and Chinese tree shrew (Tupaia chinensis) (Fig. 3; Additional file 1: Table S15 and Additional file 8: Figs. S1–S14).
Inactivation of NPB and NPBWR2 genes is found in several mammalian lineages
Sequence search and analysis for neuropeptide B (NPB) in 202 mammalian genomes returned a total of 46 species with PseudoIndex higher than 2 (Additional file 1: Table S16), the majority due to fragmentation of the genomic region (presence of Ns), true absence of exons, poor alignment identity or incompleteness of the scaffold in the NPB genomic region (Additional file 1: Table S17). From these species, members of Delphinoidea presented conserved (and validated) frameshift mutations in exon 1 (Fig. 2; Additional file 1: Table S17 and Additional file 9: Figs. S1–S7). With respect to neuropeptides B and W receptor 2 (NPBWR2), 107 species displayed a PseudoIndex higher than 2 (Additional file 1: Table S18). However, in 52 species with NPBWR2 putatively pseudogenized, such coding status was predicted due to a true absence of exons, poor alignment identity or fragmentation of the genomic region (presence of Ns) (Fig. 3; Additional file 1: Table S19). Notwithstanding, ORF-disrupting mutations were identified in 58 species mostly affecting Cetacea and Carnivora orders, but also members of Lagomorpha, Perissodactyla, Proboscidea, Primates and Rodentia orders (Fig. 3; Additional file 1: Table S19 and Additional file 10: Figs. S1–S15).
Recurrent erosion of a neuromedin, a somatostatin and a neuropeptide Y receptors in mammals
We additionally investigated the coding condition of three neuropeptide receptors given their “low-quality” tag in Cetacea genome annotations: neuromedin B receptor (NMBR), somatostatin receptor 4 (SSTR4) and neuropeptide Y receptor Y6 (Npy6r). For NMBR, 35 species displayed a PseudoIndex higher than 2 (Additional file 1: Table S20); however, validated ORF-disrupting mutations were only determined in toothed whales and golden snub-nosed monkey (Rhinopithecus roxellana) (Fig. 3; Additional file 1: Table S21 and Additional file 11: Figs. S1–S8). On the other hand, a detailed analysis of SSTR4 occurrence in mammalian genomes revealed an extensive level of gene loss in mammals, with 104 species presenting a PseudoIndex higher than 2 (Additional file 1: Table S22). In general, no ORF was detected in the genomes of cetaceans, even-toed ungulates, marine and dog-like carnivorans, bats, pangolins, horses, mole-rats, manatee and aardvark (Fig. 3; Additional file 1: Table S23 and Additional file 12: Figs. S1–S20). For Npy6r, a total of 133 species exhibited a PseudoIndex higher than 2 (Additional file 1: Table S24), with gene lesion events found and validated in most of cetaceans, even-toed ungulates, primates, carnivores, eulipotyphlans and finally in some rodents (Fig. 3; Additional file 1: Table S25 and Additional file 13: Figs. S1–S22).
RNA-seq expression
Further examination of several transcriptome datasets of four different cetacean species (Additional file 1: Table S26) allowed the assessment of the functional condition of several neuropeptides and receptors reported in this study. Priorly, assessment of the quality of the transcriptomic datasets was performed by checking transcript abundance in genes reported as being highly expressed in a specific tissue, using the Human Protein Atlas as a proxy (http://www.proteinatlas.org). Overall, we could confirm high levels of transcript abundance in the expected tissues, except in testes and brain where no/low expression was found in the expected genes (Additional file 1: Table S27).
Across all species, there was a low number of mRNA reads of our target genes, except in NPFF (for all species), NPB (for Balaenoptera acutorostrata), QRFPR (for Balaena mysticetus) and NPFFR1/NPFFR2 (for Monodon monoceros) (Additional file 1: Table S28).
After counting the number of spliced reads, exon–intron reads and exonic reads in genes presenting some gene expression, transcriptomic data presented a substantially high proportion of exon–intron reads versus spliced reads in NPFF, in stark contrast to the pattern found for QRFPR (for B. mysticetus), NPFFR1 (for Delphinapterus leucas) and NPFFR2 (for M. monoceros) (Additional file 1: Table S28). Moreover, further verification of the presence of ORF disruptive mutations in the produced transcripts allowed the detection of at least one premature stop codon (or lack of terminal stop codon as observed in NPB and NPFFR1 for Monodontidae) in the transcripts of the analysed cetacean species (Additional file 14: Figs. S1–S2; Additional file 15: Figs. S1–S2; Additional file 16: Figs. S1–S3; Additional file 17: Figs. S1–S3; Additional file 18: Fig. S1; Additional file 19: Figs. S1–S2 and Additional file 20: Figs. S1–S2) revealing either the production of truncated or extended proteins. This is especially relevant in cases where we observed a considerably distinct ratio of exon–intron reads/spliced reads among the remaining species (QRFPR for B. mysticetus, NPFFR1 for D. leucas and NPFFR2 for M. monoceros) (Additional file 1: Table S28).
Selection analyses indicates relaxation of purifying selection in NMBR of Mysticeti
To examine whether genes that are inactivated in odontocetes but not in mysticetes (QRFP, NPS, NPSR1, NPB and NMBR) exhibit a relaxation in the intensity of selection (both positive and negative), we employed the RELAX method, which explicitly incorporates a selection intensity parameter (k). With the exception of NMBR, for which the RELAX test for selection relaxation (k = 0.17) yielded significance (p = 0.01, LR = 10.38), we did not find any evidence for relaxation in the selection intensity for the other genes (Additional file 1: Table S29). Further analyses using aBSREL and BUSTED did not reveal any evidence for positive selection in the Mysticeti clade for the NMBR gene (Additional file 1: Table S30). Therefore, the observed rate of acceleration in mysticetes likely indicates a trend of relaxed purifying selection rather than an episode of positive selection.
Discussion
From an evolutionary perspective, how the neuronal control of physiological systems modifies and adjusts to radical habitat transitions is mostly unknown. Importantly, these modifications translate into considerable alterations at the anatomical (i.e. pineal gland loss [32]), physiological (i.e. hypoxia-induced adaptations [33]) and behavioural levels (i.e. sleep [34]). Still, the molecular basis of such changes—in perception, motor control, sleep, spatial control, motivation and learning/memory—is challenging to assess. Here, we used as proxy the plethora of neuropeptides known to regulate a vast network of functions: including sleep, feeding, pain, stress, immunity (Fig. 1). Some of the reported receptors have previously been identified as lost in early vertebrate lineages, such as SSTR4 in ray-finned fish [35], or in the ancestors of mammals and birds, like QRFPR [36]. Additionally, certain receptors, such as NPFFR2 [37], QRFPR [5], SSTR4 [35] and Npy6r [38], have been reported as lost in other mammals.
Comparative analysis of 40 cetacean genomes uncovered an extensive loss pattern of several neuropeptides and receptors (Figs. 2 and 3). The marked gene loss pattern of pleiotropic actors (e.g. [39]), acting on different tissues, anticipates the drastic modification of distinct gene regulatory networks as a result of adaptation to a fully aquatic lifestyle. However, regarding some neuropeptides and receptors (e.g. NPB), Mysticeti (baleen whales) seem to retain functional genes (Fig. 2). A similar outcome was previously observed in other organ systems (e.g. [40]), thus implying the existence of critical changes in selective pressures over the time in these two related lineages. Nevertheless, our results also suggest that the intensity of purifying selection acting on Mysticeti NMBR has been relaxed, which might indicate earlier stages of gene inactivation. Therefore, at least for NMBR, it is possible that loss of function has occurred on the stem cetacean branch, rather than on the branch leading to the radiation of odontocetes.
Circadian rhythmicity is one of the most prominent behavioural adaptations in Cetacea. A strong association between the loss of the full melatonin-related gene hub and the existence of unique bio-rhythmicity in cetaceans has been described [5, 7, 9]. Moreover, disruption of neuropeptides and receptors, such as the neuropeptide cortistatin and the dopamine receptor D5 (DRD5), has been correlated with changes in daily activity patterns and energy metabolism [8, 41]. Thus, the absence of a subset of neuropeptides and receptors may have consequences in terms of the neuroendocrine regulation of Cetacea circadian rhythmicity and sleeping behaviour. In agreement, some of the analysed receptors are expressed in hypocretin/orexin neurons (i.e. NPFFRs) [42] and QRFP was suggested to partially co-localize with orexin [43]. The orexinergic system plays a crucial role in regulating sleep/wake rhythms and thermoregulation, but also in cardiovascular responses, feeding behaviour, spontaneous physical activity and control of energy metabolism [44]. Given the differences in terms of orexinergic bouton density [10] and number of hypothalamic orexinergic neurons [45] found in Cetacea in comparison with their closest relatives (even-toed ungulates), it is expected that disruption of these endocrine messengers may influence cetacean’s unique behaviour. Strikingly, we report the loss of sleep inducing (QRFP, NPB) [17, 19, 22] or arousal (NPS) [24, 26] neuropeptides, and related receptors, in most Cetacea. Yet, sleep-modulators somatostatin (SST) and neuropeptide Y (NPY) show no signs of erosion [46, 47]. In Cetacea, the loss of multiple neuropeptide sleep regulation seems to have paired with a novel rearrangement of sleep: with the loss of REM sleep and the transition from bihemispheric into unihemispheric sleep [34]. Such transition likely entailed novel regulatory networks acting on a single brain hemisphere rather than the brain as a whole. In agreement, the emergence of Cetacea unihemispheric sleep was accompanied by local thermoregulatory adjustments: uncoupling the brain hemispheric temperature and stabilizing brainstem temperature to prevent sleep-inducing cooling and maintaining motor and sensorial functions (Fig. 4) [48, 49]. In addition, hemisphere communication was also adjusted, with the occurrence of interhemispheric fascicle projections, directly into dorsal thalamus nuclei instead of the brainstem projections observed in other mammals [34]. Such functional and anatomical changes possibly allowed the simultaneous vigilance and sleep needed for a mammal to inhabit the high sea.

Loss of neuropeptides and receptors contributed to adaptation in an aquatic environment. Impairment of neuropeptides and receptors led to drastic modifications in terms of response to physiological stress, biological rhythmicity and metabolism
A signalling shift seems to have also occurred in the context of the Cetacea diving response: during which the heart rate is reduced (bradycardia) and the consequent drop in arterial blood pressure is restrained through a strategy of peripheral vasoconstriction, to maintain blood distribution to heart and brain during diving apnoea [50]. Previous reports associated events of gene loss and adaptive gene evolution (positive selection) to the modulation of Cetacea blood volume, pressure and peripheral vasoconstriction [41, 51, 52]. The loss of the hypertensive and heart rate-modulating neuropeptides and receptors (NPFF, QRFP, NPB; cognate receptors and NMBR) seems to further contribute to the blood pressure and heart rate regulation [16, 17, 53,54,55,56]. NPFF in particular was shown to be responsible, in rat (Rattus norvegicus), for up to 50% of the cardiac component of the baroreflex, participating in the maintenance of the blood pressure levels, and to modulate the chemoreflex through action on ASIC channels, which elicits hyperventilation [16, 53, 57,58,59,60]. SSTR4 was also suggested to modulate ventilation, with airway contraction observed in knock-out mice [61]. On the other hand, the intact neuropeptides SST and NPY were suggested to modulate breathing, notably under hypoxic conditions, with SST attenuating ventilation and inducing apnoea responses [62, 63]. NPY is also known for its prominent effects on the cardiovascular system [64]; thus, conservation of NPY may importantly contribute to blood pressure regulation in this group.
Metabolism is yet another major target of neuropeptide regulation through feeding behaviour and energy expenditure (NPFF; NPB/NPW; QRFP; NMBR) [17, 20, 54, 65,66,67] or by controlling insulin secretion (QRFP; NPB/NPW) [18, 68, 69]. Accordingly, cetaceans are known to present unique metabolic needs to adapt to a low glucose diet and to the metabolic constraints of oxygen restriction (Fig. 4) [70, 71]. Thus, loss of these feeding suppression neuropeptides and metabolic modulators could contribute to fat signalling and deposition, as well as to the peripheral insulin resistance and high circulating glucose levels under fasting, as observed in odontocete cetaceans [72, 73]. These conditions seem to ensure a steady glucose supply to the central nervous system in the absence of alternative fuel sources—i.e. ketogenesis impairment resulting from genomic loss of hydroxymethyl-CoA synthase—in the context of their low carbohydrate, high protein and fat diet (Fig. 4) [74,75,76]. In agreement, NPY, an inducer of obesity and fat storage, was found intact, possibly contributing to the overall fat profile in Cetacea [77]. Although fat deposition and obesity trigger adipose tissue inflammation in most mammals, Cetacea exhibit a healthy fat phenotype [70, 78]. This could be concurrent with the loss of inflammatory modulation by NPFF, NPS or NPB/NPW [15, 79, 80]. In addition to adipose tissue inflammation, the loss of these neuropeptides and receptors, notably the SST receptor SSTR4 or NPBWR2 [23, 61], might have further helped cetaceans extend their diving capacities by attenuating lung pain and inflammation, resulting from lung collapse as well as the formation of gas bubbles during deeper dives [52, 80,81,82,83].
Interestingly, we observed a lack of conserved founding mutations common to all analysed Cetacea in most study genes, thus indicating that pseudogenization events took place after the diversification of modern Cetacea lineages (Fig. 2). Given the role of neuropeptides as master regulators of a different set of physiological processes, it is expected that, in the course of evolution, molecules with a pleiotropic action should suffer a progressive decay, in parallel with the modifications of the physiological compartments in which they are associated. Adaptation to specific ecological niches might have triggered the occurrence of disrupting mutations in different genomic positions/timescales, thus promoting independent pseudogenization events.
Our approach also identified evolutionary convergence in patterns of gene loss across marine mammals, NPS and NPSR1 (Fig. 3). Together, these results suggest that these convergent genomic variations possibly contributed to the transition from land to water in marine mammal lineages. This is a pattern previously reported for other genes [5, 84, 85] and reinforces the idea of gene loss as a remarkable molecular signature of adaptive evolution in habitat shifts. We also have found other convergent patterns of disruption less easy to explain from an evolutionary perspective. As an example, convergent inactivation of the NPFF and receptors (NPFF and NPFFR2 as in Cetacea results on NPFFR1 coding status were not completely clear) and QRFP and receptor in flying foxes (Pteropus sp.) and Cetaceans (Fig. 3) might be a genomic signature of adaptation to novel niches—an evolutionary convergence previously reported for other genes (e.g. [86]).
Conclusions
Our findings suggest that alteration/loss of neuropeptide and receptors parallel adaptive shifts in behavioural processes such as bio-rhythmicity, diving and feeding behaviour. On another level, the patterns of gene modification observed in this study reinforce the idea of gene loss as a major evolutionary driver in the land-to-water habitat transition experienced by the Cetacea ancestor.
Methods
Genomic sequence compilation
An exhaustive literature review was used to identify a list of key genes that encode neuropeptides and respective receptors with pleiotropic action in physiological compartments that have suffered drastic adjustments in the land to water transition. Consequently, NPFF, NPFFR1, NPFFR2, QRFP, QRFPR, NPS, NPSR1, NPB, NPBWR2, NMBR, SSTR4 and Npy6r corresponding genomic regions from 172 mammals with the genome annotated were collected from NCBI (Additional file 1: Table S1). Retrieval was performed using Genomic Sequence Downloader (https://github.com/luisqalves/genomic_sequence_downloader.py), using human synteny for each target gene as input—downstream and upstream flanking genes considered were the ones presenting a gene type as “protein coding”. Whenever Genomic Sequence Downloader failed to obtain a genomic region containing the target gene, there was a tempt to collect directly the corresponding genomic sequences from the reference genome assemblies available at NCBI—either using the target gene or the corresponding flanking genes to tell us the genomic region comprising the physical location of the gene. In addition to the species previously referred, 29 genomes from cetaceans with no annotated genome available and Hippopotamus amphibius (hippopotamus)—either from NCBI, DNA Zoo [87] or Bowhead Whale Genome Resource (http://www.bowhead-whale.org)—were also inspected. In this case, the available genome assemblies were searched through blastn using Bos taurus target gene corresponding orthologue coding sequence (CDS), as well as the CDSs of the flanking genes in the same species. Exception made for SSTR4 where Homo sapiens CDS for target gene and flanking genes were used as query. The best matching genome scaffold was retrieved. When no consensual blast hit was obtained, all hits corresponding to the query were inspected, the aligning regions submitted to a back-blast search against the nucleotide (nt) database of NCBI, with the matching genomic sequence(s) corresponding to the gene of interest being the one(s) selected for annotation (when existing).
Inference of the coding status
Gene annotation was firstly performed using a pseudogene inference pipeline, PseudoChecker [31]. For each run, the Homo sapiens (human) orthologous gene and genomic sequences were used as reference (NCBI Accession ID regarding human NPFF: NM_003717.4; NPFFR1: NM_022146.5; NPFFR2: NM_004885.3; QRPF: NM_198180.3; QRPFR: NM_198179.3; NPS: NM_001030013.2; NPSR1: NM_207172.2; NPB: NM_148896.5; NPBWR2: NM_005286.4; NMBR: NM_002511.4; SSTR4: NM_001052.4; default parameters were maintained). For gene annotation in cetaceans and hippopotamus, B. taurus CDS were USED as reference, whenever it presented a curated transcript (NCBI Accession ID regarding cattle NPFF: NM_174123.3; QRFP: NM_198222.1; QRFPR: NM_001192681.1; NPSR1: NM_001192977.2; NPB: NM_173944.1; NPBWR2: NM_174075.1; NMBR: NM_001205710.1). Finally, for Npy6r annotation, Mus musculus (house mouse) orthologous gene was used as reference (NCBI Accession ID: NM_010935.4). Estimation of the erosion condition of the tested genes was performed through PseudoIndex, a user assistant metric built into the software PseudoChecker, that ranges on a discrete scale from 0 (coding) to 5 (pseudogenized) [31]. PseudoIndex considers three key components: absent exons, shifted codons and truncated sequences, each quantifying various aspects of mutational evidence. The predicted sequences were classified into “functional” if PseudoIndex was between 0 and 2, or “putatively pseudogenized” when PseudoIndex was higher than 2. Manual gene annotation and validation was performed for species presenting PseudoIndex higher than 2, with the collected genomic sequences being uploaded into Geneious Prime® 2021.2.2 and the gene sequence manually predicted as described in Lopes-Marques [88] using the previous coding sequences from the same species as reference. Briefly, reference exons were mapped to the corresponding genomic sequences and subsequently aligned regions were manually inspected to find putatively ORF disrupting mutations (frameshifts (indels), premature stop codon, loss of canonical splice sites). Given the small size of the first exon in human NPFFR1 and NPS (only 7 and 8 base pairs, respectively), putative inactivating mutations in these exons were not considered. The identified ORF disrupting mutations (one per species) were validated by searching at least two independent Sequence Read Archive (SRA) projects (when possible) of the corresponding species. Conserved deleterious mutations were validated in only one species.
Assessment of gene expression in multiple tissues
The global analyses of relative gene expression in Cetacea species were performed in four cetacean species, using the approach described previously [89]. The genome and annotations of D. leucas (Accession number: GCF_002288925.2), M. monoceros (Accession number: GCF_005190385.1) and B. acutorostrata (Accession number: GCF_000493695.1) were downloaded from NCBI genome browser. On the other hand, the genome and annotation of B. mysticetus were retrieved from http://www.bowhead-whale.org/. Next, we collected the RNA-seq datasets from NCBI (Additional file 1: Table S26). When more than one dataset was available for a specific tissue, both were concatenated. Before proceeding with relative gene expression analyses, the genomes and annotations were checked for errors with (agat_convert_sp_gxf2gxf.pl) script and converted from.gff to.gtf format with (agat_convert_sp_gff2gtf.pl) script of AGAT tool [90]. The RNA-seq datasets were mapped against each genome with the Hisat2 v.2.2.1 aligner [91, 92], and the relative gene expression determined with the StingTie v.2.2.1 software [93], following the authors protocol [94]. In the end, the gene expression quantifications were used in transcript per million (TPM).
To assess the quality of each transcriptomic dataset, we calculated gene expression in a highly expressed gene according to the Human Protein Atlas (http://www.proteinatlas.org). Genes included in this analysis were the following: arginine vasopressin (AVP; for brain), phosducin (PDC; for retina), uromodulin (UMOD; for kidney), transition protein-1 (TNP1; for testes), nicotinamide riboside kinase 2 (NMRK2; for muscle), acrosin (ACR; for heart), secreted phosphoprotein 2 (SPP2; for liver), advanced glycosylation end-product specific receptor (AGER; for lung), C–C motif chemokine ligand 25 (CCL25; for lymph) and leptin (LEP; for blubber).
To inspect the occurrence of expression reads from our target genes, mRNA reads previously collected were mapped to corresponding annotated genes using the “map to reference” tool available in Geneious Prime® 2021.2.2 (maximum read gap size adjusted to the length of the maximum distance between exons with a maximum mismatch rate of 1%). The mapped reads were then classified as either spliced reads (spanning over two exons), exon–intron reads or exonic reads, based on the genomic region they mapped to. After classification, the reads of each class were counted, and aligning regions covering mutated regions were screened for mutated transcripts.
Molecular evolutionary analyses
Given the slow rate of evolution in Mysticeti [95], evidence for pseudogenization does not accumulate rapidly. For neuropeptide and receptor genes that undergo knockout in odontocetes, yet remain intact in mysticetes (QRFP, NPS, NPSR1, NPB, NMBR), estimation of the selection intensity acting on these genes (through dN/dS analyses) within Mysticeti would provide valuable insights. For this purpose, we translation-aligned the predicted sequences of Mysticeti (retrieved from PseudoChecker) and several mammals from different lineages without any inactivating mutations (see Additional file 1: Table S31) using the Blosum62 substitution matrix in Geneious Prime® 2021.2.2. To investigate potential relaxation of purifying selection, often linked with gene pseudogenization, we conducted RELAX analysis using the HyPhy package [96, 97]. This analysis involves comparing a foreground set of species (Mysticeti in this case) with a background set (all non-cetacean species) within a hypothesis-testing framework. In cases where significant evidence of relaxation was found, we additionally run aBSREL [98] and BUSTED [99] to ascertain whether positive selection has manifested across a subset of branches or to assess gene-wide (rather than site-specific) positive selection. The idea behind these analyses is that if there is no evidence of positive selection, the accelerated rate in a particular clade probably indicates a pattern of relaxed purifying selection.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. The data that support the findings of this study are openly available in the National Center for Biotechnology Information (NCBI) Assembly database (https://www.ncbi.nlm.nih.gov/assembly/), as well as NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra/). Genome assemblies are also available at DNA Zoo (https://www.dnazoo.org/) and at The Bowhead Whale Genome Resource (http://www.bowhead-whale.org/). The specific accession numbers (i.e. genome assemblies, scaffold IDs and SRA and sample IDs) for all data used from these publicly available databases can be found in the supplementary information files.
Abbreviations
- NPFF :
-
Neuropeptide FF-amide peptide precursor
- NPFFR1 :
-
Neuropeptide FF receptor 1
- NPFFR2 :
-
Neuropeptide FF receptor 2
- QRFP :
-
Pyroglutamylated RFamide peptide
- QRFPR :
-
Pyroglutamylated RFamide peptide receptor
- NPS :
-
Neuropeptide S
- NPSR1 :
-
Neuropeptide S receptor 1
- NPB :
-
Neuropeptide B
- NPBWR2 :
-
Neuropeptides B and W receptor 2
- NMBR :
-
Neuromedin B receptor
- SSTR4 :
-
Somatostatin receptor 4
- Npy6r :
-
Neuropeptide Y receptor Y6
- ORF:
-
Open reading frame
- AVP :
-
Arginine vasopressin
- PDC :
-
Phosducin
- UMOD :
-
Uromodulin
- TNP1 :
-
Transition protein-1
- NMRK2 :
-
Nicotinamide riboside kinase 2
- ACR :
-
Acrosin
- SPP2 :
-
Secreted phosphoprotein 2
- AGER :
-
Advanced glycosylation end-product specific receptor
- CCL25 :
-
C-C motif chemokine ligand 25
- LEP :
-
Leptin
- mRNA:
-
Messenger RNA
- TPM:
-
Transcripts per million
- SRA:
-
Sequence Read Archive
- CDS:
-
Coding sequence
- RNA-seq:
-
RNA-sequencing
- GPCR:
-
G protein-coupled receptor
- DRD5 :
-
Dopamine receptor D5
- NPY :
-
Neuropeptide Y
- REM:
-
Rapid eye movement
- SST :
-
Somatostatin
- NPW :
-
Neuropeptide W
- dN/dS:
-
Ratio of non-synonymous to synonymous substitutions
References
Bishop KL. The evolution of flight in bats: narrowing the field of plausible hypotheses. Q Rev Biol. 2008;83:153–69.
Graham LE, Cook ME, Busse JS. The origin of plants: body plan changes contributing to a major evolutionary radiation. Proc Natl Acad Sci USA. 2000;97:4535–40.
Hu Y, Wang X, Xu Y, Yang H, Tong Z, Tian R, et al. Molecular mechanisms of adaptive evolution in wild animals and plants. Sci China Life Sci. 2023;13:1–43.
McGowen MR, Gatesy J, Wildman DE. Molecular evolution tracks macroevolutionary transitions in Cetacea. Trends Ecol Evol. 2014;296:336–46.
Huelsmann M, Hecker N, Springer MS, Gatesy J, Sharma V, Hiller M. Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Sci Adv. 2019;5:eaaw6671.
Themudo GE, Alves LQ, Machado AM, Lopes-Marques M, da Fonseca RR, Fonseca M, et al. Losing genes: the evolutionary remodeling of Cetacea skin. Front Mar Sci. 2020;7: 592735.
Lopes-Marques M, Ruivo R, Alves LQ, Sousa N, Machado AM, Castro LFC. The singularity of Cetacea behavior parallels the complete inactivation of melatonin gene modules. Genes. 2019;10:121.
Valente R, Alves LQ, Nabais M, Alves F, Sousa-Pinto I, Ruivo R, et al. Convergent cortistatin losses parallel modifications in circadian rhythmicity and energy homeostasis in Cetacea and other mammalian lineages. Genomics. 2021;113:1064–70.
Emerling CA, Springer MS, Gatesy J, Jones Z, Hamilton D, Xia-Zhu D, et al. Genomic evidence for the parallel regression of melatonin synthesis and signaling pathways in placental mammals. Open Res Eur. 2021;1:75. https://doi.org/10.12688/openreseurope.13795.1.
Dell L-A, Spocter MA, Patzke N, Karlson KÆ, Alagaili AN, Bennett NC, et al. Orexinergic bouton density is lower in the cerebral cortex of cetaceans compared to artiodactyls. J Chem Neuroanat. 2015;68:61–76.
Hökfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides - an overview. Neuropharmacology. 2000;398:1337–56.
Strand F. Neuropeptides: regulators of physiological processes. Cambridge: MIT Press; 1999.
Berta A, Sumich JL, Kovacs KM. Marine mammals evolutionary biology. United States: Academic Press; 2015.
Roumy M, Zajac JM. Neuropeptide FF, pain and analgesia. Eur J Pharmacol. 1998;345:1–11.
Panula P, Kalso E, Nieminen M-L, Kontinen VK, Brandt A, Pertovaara A. Neuropeptide FF and modulation of pain. Brain Res. 1999;848:191–6.
Fang Q, Li N, Jiang TN, Liu Q, Li YL, Wang R. Pressor and tachycardic responses to intrathecal administration of neuropeptide FF in anesthetized rats. Peptides. 2010;31:683–8.
Takayasu S, Sakurai T, Iwasaki S, Teranishi H, Yamanaka A, Williams SC, et al. A neuropeptide ligand of the G protein-coupled receptor GPR103 regulates feeding, behavioural arousal, and blood pressure in mice. Proc Natl Acad Sci USA. 2006;103:7438–43.
Chartrel N, Picot M, El Medhi M, Arabo A, Berrahmoune H, Alexandre D, et al. The neuropeptide 26RFa (QRFP) and its role in the regulation of energy homeostasis: a mini-review. Front Neurosci. 2016;10:549.
Chen A, Chiu CN, Mosser EA, Kahn S, Spence R, Prober DA. QRFP and its receptors regulate locomotor activity and sleep in zebrafish. J Neurosci. 2016;36:1823–40.
Tanaka H, Yoshida T, Miyamoto N, Motoike T, Kurosu H, Shibata K, et al. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc Natl Acad Sci USA. 2003;100:6251–6.
Samson WK, Baker JR, Samson CK, Samson HW, Taylor MM. Central neuropeptide B administration activates stress hormone secretion and stimulates feeding in male rats. J Neuroendocrinol. 2004;16:842–9.
Hirashima N, Tsunematsu T, Ichiki K, Tanaka H, Kilduff TS, Yamanaka A. Neuropeptide B induces slow wave sleep in mice. Sleep. 2011;34:31–7.
Sakurai T. NPBWR1 and NPBWR2: implications in energy homeostasis, pain, and emotion. Front Endocrinol. 2013;4:23.
Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, et al. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–97.
Smith KL, Patterson M, Dhillo WS, Patel SR, Semjonous NM, Gardiner JV, et al. Neuropeptide S stimulates the hypothalamo-pituitary-adrenal axis and inhibits food intake. Endocrinology. 2006;147:3510–8.
Xing L, Shi G, Mostovoy Y, Gentry NW, Fan Z, McMahon TB, et al. Mutant neuropeptide S receptor reduces sleep duration with preserved memory consolidation. Sci Transl Med. 2019;11:eaax2014.
Oliveira KJ, Cabanelas A, Veiga MA, Paula GS, Ortiga-Carvalho TM, Wada E, et al. Impaired serum thyrotropin response to hypothyroidism in mice with disruption of neuromedin B receptor. Regul Pept. 2008;146:213–7.
Yang G, Huang H, Tang M, Cai Z, Huang C, Qi B, et al. Role of neuromedin B and its receptor in the innate immune responses against influenza A virus infection in vitro and in vivo. Vet Res. 2019;50:80.
Scheich B, Csekő K, Borbély É, Ábrahám I, Csernus V, Gaszner B, et al. Higher susceptibility of somatostatin 4 receptor gene-deleted mice to chronic stress-induced behavioral and neuroendocrine alterations. Neuroscience. 2017;346:320–36.
Yulyaningsih E, Loh K, Lin S, Lau J, Zhang L, Shi Y, et al. Pancreatic polypeptide controls energy homeostasis via Npy6r signaling in the suprachiasmatic nucleus in mice. Cell Metab. 2014;19:58–72.
Alves LQ, Ruivo R, Fonseca MM, Lopes-Marques M, Ribeiro P, Castro LFC. PseudoChecker: an integrated online platform for gene inactivation inference. Nucleic Acids Res. 2020;48:W321–31.
Ralph CL. The pineal gland and geographical distribution of animals. Int J Biometeorol. 1975;19:289–303.
Tian R, Yin D, Liu Y, Seim I, Xu S, Yang G. Adaptive evolution of energy metabolism-related genes in hypoxia-tolerant mammals. Front Genet. 2017;8:205.
Lyamin OI, Manger PR, Ridgway SH, Mukhametov LM, Siegel JM. Cetacean sleep: an unusual form of mammalian sleep. Neurosci Biobehav Rev. 2008;32:1451–84.
Ocampo Daza D, Sundström G, Bergqvist CA, Larhammar D. The evolution of vertebrate somatostatin receptors and their gene regions involves extensive chromosomal rearrangements. BMC Evol Biol. 2012;12:231–310.
Larhammar D, Xu B, Bergqvist CA. Unexpected multiplicity of QRFP receptors in early vertebrate evolution. Front Neurosci. 2014;8:337.
LeDuc D, Velluva A, Cassat-Johnstone M, Olsen RA, Baleka S, Lin C-C, et al. Genomic basis for skin phenotype and cold adaptation in the extinct Steller’s sea cow. Sci Adv. 2022;8:eabl6496.
Starbäck P, Wraith A, Eriksson H, Larhammar D. Neuropeptide Y receptor gene y6: multiple deaths or resurrections? Biochem Biophys Res Commun. 2000;277:264–9.
Panula P, Aarnisalo AA, Wasowicz K. Neuropeptide FF, a mammalian neuropeptide with multiple functions. Prog Neurobiol. 1996;48:461–87.
Alves LQ, Ruivo R, Valente R, Fonseca MM, Machado AM, Plön S, et al. A drastic shift in the energetic landscape of toothed whale sperm cells. Curr Biol. 2021;31:3648–55.
Alves LQ, Alves J, Ribeiro R, Ruivo R, Castro LFC. The dopamine receptor D5 gene shows signs of independent erosion in toothed and baleen whales. PeerJ. 2019;7: e7758.
Li S-B, Giardino WJ, de Lecea L. Hypocretins and arousal. Curr Top Behav Neurosci. 2017;33:93–4.
Sagi D, de Lecea L, Appelbaum L. Heterogeneity of hypocretin/orexin neurons. Front Neurol Neurosci. 2021;45:61–74.
Chieffi S, Carotenuto M, Monda V, Valenzano A, Villano I, Precenzano F, et al. Orexin system: the key for a healthy life. Front Physiol. 2017;8:357.
Dell L-A, Patzke N, Bhagwandin A, Bux F, Fuxe K, Barber G, et al. Organization and number of orexinergic neurons in the hypothalamus of two species of Cetartiodactyla: a comparison of giraffe (Giraffa camelopardalis) and harbour porpoise (Phocoena phocoena). J Chem Neuroanat. 2012;44:98–9.
Obál F Jr, Krueger JM. The somatotropic axis and sleep. Rev Neurol. 2001;157:S12-15.
Singh C, Rihel J, Prober DA. Neuropeptide Y regulates sleep by modulating noradrenergic signaling. Curr Biol. 2017;27:3796–811.
Kovalzon VM, Mukhametov LM. Temperature fluctuations of the dolphin brain corresponding to unihemispheric slow-wave sleep. J Evol Biochem Physiol. 1983;18:222–4.
Siegel JM. Sleep function: an evolutionary perspective. Lancet Neurol. 2022;21:937–46.
Panneton WM. The mammalian diving response: an enigmatic reflex to preserve life? Physiology. 2013;28:284–97.
Tian R, Wang Z, Niu X, Zhou K, Xu S, Yang G. Evolutionary genetics of hypoxia tolerance in cetaceans during diving. Genome Biol Evol. 2016;8:827–39.
Ding X, Yu F, He X, Xu S, Yang G, Ren W. Rubbing salt in the wound: molecular evolutionary analysis of pain-related genes reveals the pain adaptation of cetaceans in seawater. Animals. 2022;12:3571.
Laguzzi R, Nosjean A, Mazarguil H, Allard M. Cardiovascular effects induced by the stimulation of neuropeptide FF receptors in the dorsal vagal complex: an autoradiographic and pharmacological study in the rat. Brain Res. 1996;711:193–202.
Ohki-Hamazaki H, Neuromedin B. Prog Neurobiol. 2000;62:297–312.
Yu N, Chu C, Kunitake T, Kato K, Nakazato M, Kannan H. Cardiovascular actions of central neuropeptide W in conscious rats. Regul Pept. 2007;138:82–6.
Dvorakova CM. Distribution and function of neuropeptides W/B signaling system. Front Physiol. 2018;9:981.
Somers VK, Mark AL, Zavala DC, Abboud FM. Influence of ventilation and hypocapnia on sympathetic nerve responses to hypoxia in normal humans. J Appl Physiol. 1989;67:2095–100.
Askwith CC, Cheng C, Ikuma M, Benson C, Price MP, Welsh MJ. Neuropeptide FF and FMRFamide potentiate acid-evoked currents from sensory neurons and proton-gated DEG/ENaC channels. Neuron. 2000;26:133–41.
Catarsi S, Babinski K, Seguela P. Selective modulation of heteromeric ASIC proton-gated channels by neuropeptide FF. Neuropharmacology. 2001;41:592–600.
Jhamandas JH, Goncharuk V. Role of neuropeptide FF in central cardiovascular and neuroendocrine regulation. Front Endocrinol. 2013;4:8.
Helyes Z, Pintér E, Sándor K, Elekes K, Bánvölgyi A, Keszthelyi D, et al. Impaired defense mechanism against inflammation, hyperalgesia, and airway hyperreactivity in somatostatin 4 receptor gene-deleted mice. Proc Natl Acad Sci USA. 2009;106:13088–93.
Maxwell DL, Chahal P, Nolop KB, Hughes JMB. Somatostatin inhibits the ventilatory response to hypoxia in humans. J Appl Physiol. 1986;60:997–1002.
Corder R, Pralong FP, Muller AF, Gaillard RC. Regional distribution of neuropeptide Y-like immunoreactivity in human hypothalamus measured by immunoradiometric assay: possible influence of chronic respiratory failure on tissue levels. Neuroendocrinology. 1990;51:23–30.
Walker P, Grouzmann E, Burnier M, Waeber B. The role of neuropeptide Y in cardiovascular regulation. Trends Pharmacol Sci. 1991;12:111–5.
Murase T, Arima H, Kondo K, Oiso Y. Neuropeptide FF reduces food intake in rats. Peptides. 1996;17:353–4.
Sunter D, Hewson AK, Lynam S, Dickson SL. Intracerebroventricular injection of neuropeptide FF, an opioid modulating neuropeptide, acutely reduces food intake and stimulates water intake in the rat. Neurosci Lett. 2001;313:145–8.
Mondal MS, Yamaguchi H, Date Y, Shimbara T, Toshinai K, Shimomura Y, et al. A role for neuropeptide W in the regulation of feeding behavior. Endocrinology. 2003;144:4729–33.
Dezaki K, Kageyama H, Seki M, Shioda S, Yada T. Neuropeptide W in the rat pancreas: potentiation of glucose-induced insulin release and Ca2+ influx through L-type Ca2+ channels in beta-cells and localization in islets. Regul Pept. 2008;145:153–8.
Billert M, Sassek M, Wojciechowicz T, Jasaszwili M, Strowski MZ, Nowak KW, et al. Neuropeptide B stimulates insulin secretion and expression but not proliferation in rat insulin-producing INS-1E cells. Mol Med Rep. 2019;20:2030–8.
Wang Z, Chen Z, Xu S, Ren W, Zhou K, Yang G. ‘Obesity’ is healthy for cetaceans? Evidence from pervasive positive selection in genes related to triacylglycerol metabolism. Sci Rep. 2015;5:14187.
Derous D, Sahu J, Douglas A, Lusseau D, Wenzel M. Comparative genomics of cetartiodactyla: energy metabolism underpins the transition to an aquatic lifestyle. Conserv Physiol. 2021;9: a136.
Venn-Watson SK, Ridgway SH. Big brains and blood glucose: common ground for diabetes mellitus in humans and healthy dolphins. Comp Med. 2007;57:390–5.
Venn-Watson S, Carlin K, Ridgway S. Dolphins as animal models for type 2 diabetes: sustained, post-prandial hyperglycemia and hyperinsulinemia. Gen Comp Endocrinol. 2011;170:193–9.
Jebb D, Hiller M. Recurrent loss of HMGCS2 shows that ketogenesis is not essential for the evolution of large mammalian brains. eLife. 2018;7:e38906.
Houser DS, Derous D, Douglas A, Lusseau D. Metabolic response of dolphins to short-term fasting reveals physiological changes that differ from the traditional fasting model. J Exp Biol. 2021;224:jeb238915.
Wolfgang MJ, Choi J, Scafidi S. Functional loss of ketogenesis in odontocete cetaceans. J Exp Biol. 2021;224:jeb243062.
Vähätalo LH, Ruohonen ST, Mäkelä S, Kovalainen M, Huotari A, Mäkelä KA, et al. Neuropeptide Y in the noradrenergic neurones induces obesity and inhibits sympathetic tone in mice. Acta Physiol. 2015;213:902–19.
Hildebrandt X, Ibrahim M, Peltzer N. Cell death and inflammation during obesity: “know my methods, WAT(son).” Cell Death Differ. 2023;30:1–14.
Bernier V, Stocco R, Bogusky MJ, Joyce JG, Parachoniak C, Grenier K, et al. Structure-function relationships in the neuropeptide S receptor: molecular consequences of the asthma-associated mutation N107I. J Biol Chem. 2006;281:24704–12.
Hondo M, Ishii M, Sakurai T. The NPB/NPW neuropeptide system and its role in regulating energy homeostasis, pain, and emotion. Results Probl Cell Differ. 2008;46:239–56.
Pintér E, Helyes Z, Szolcsanyi J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol Ther. 2006;112:440–56.
Mishra SK, Holzman S, Hoon MA. A nociceptive signaling role for neuromedin B. J Neurosci. 2012;32:8686–95.
Fahlman A, Moore MJ, Wells RS. How do marine mammals manage and usually avoid gas emboli formation and gas embolic pathology? Critical clues from studies of Wild Dolphins. Front Mar Sci. 2021;8: 598633.
Chikina M, Robinson JD, Clark NL. Hundreds of genes experienced convergent shifts in selective pressure in marine mammals. Mol Biol Evol. 2016;33:2182–92.
Meyer WK, Jamison J, Richter R, Woods SE, Partha R, Kowalczyk A, et al. Ancient convergent losses of paraoxonase 1 yield potential risks for modern marine mammals. Science. 2018;3616402:591–4.
Liu Y, Cotton JA, Shen B, Han X, Rossiter SJ, Zhang S. Convergent sequence evolution between echolocating bats and dolphins. Curr Biol. 2010;20:R53–4.
Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–5.
Lopes-Marques M, Ruivo R, Fonseca E, Teixeira A, Castro LFC. Unusual loss of chymosin in mammalian lineages parallels neo-natal immune transfer strategies. Mol Phylogenet Evol. 2017;116:78–86.
Páscoa I, Fonseca E, Ferraz R, Machado AM, Conrado F, Ruivo R, et al. The preservation of PPARγ genome duplicates in some teleost lineages: insights into lipid metabolism and xenobiotic exploitation. Genes. 2022;13:107.
Dainat J, Hereñú D, Davis E, Crouch K, LucileSol, Agostinho N, et al. NBISweden/AGAT: AGAT-v1.0.0 (v1.0.0). Zenodo. 2022. https://doi.org/10.5281/zenodo.7255559
Kim D, Langmead B, Salzberg S. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Pertea M, Kim D, Pertea G, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT. StringTie and Ballgown Nat Protoc. 2016;11:1650–67.
Jackson JA, Baker CS, Vant M, Steel DS, Medrano-González L, Palumbi SR. Big and slow: phylogenetic estimates of molecular evolution in baleen whales (suborder Mysticeti). Mol Biol Evol. 2009;26:2427–40.
Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–9.
Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol. 2015;32(3):820–32.
Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol. 2015;32:1342–53.
Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, et al. Gene-wide identification of episodic selection. Mol Biol Evol. 2015;32:1365–71.
Williams EM. Synopsis of the earliest cetaceans. In: Thewissen JGM, editor. The emergence of whales. advances in vertebrate paleobiology, vol 1. Boston: Springer; 1998. p. 1–28. https://doi.org/10.1007/978-1-4899-0159-0_1.
Bajpai S, Thewissen JGM, Sahni A. The origin and early evolution of whales: macroevolution documented on the Indian subcontinent. J Biol Sci. 2009;34:673–86.
Gingerich PD, Ul-Haq M, von Koenigswald W, Sanders WJ, Smith BH, Zalmout IS. New protocetid whale from the Middle Eocene of Pakistan: birth on land, precocial development, and sexual dimorphism. PLoS ONE. 2009;4: e4366.
Spaulding M, O’leary MA, Gatesy J. Relationships of Cetacea (Artiodactyla) among mammals: increased taxon sampling alters interpretations of key fossils and character evolution. PLoS One. 2009;4:e7062.
Cooper LN, Thewissen JGM, Bajpai S, Tiwari BN. Postcranial morphology and locomotion of the Eocene raoellid Indohyus (Artiodactyla: Mammalia). Hist Biol. 2012;24:279–310.
Orliac MJ, Benoit J, O’Leary MA. The inner ear of Diacodexis, the oldest artiodactyl mammal. J Anat. 2012;221:417–26.
McQuate S. Fossil of oldest known baleen-whale relative unearthed in Peru. Nat Publ Group. 2017. https://doi.org/10.1038/nature.2017.21966.
Gingerich PD, Amane A, Zouhri S. Skull and partial skeleton of a new pachycetine genus (Cetacea, Basilosauridae) from the Aridal Formation, Bartonian middle Eocene, of southwestern Morocco. PLoS ONE. 2022;17: e0276110.
Unger RH, Dobbs RE, Orci L. Insulin, glucagons, and somatostatin secretion in the regulation of metabolism. Annu Rev Physiol. 1978;40:307–43.
Michalkiewicz M, Michalkiewicz T, Kreulen DL, McDougall SJ. Increased blood pressure in neuropeptide Y transgenic rats. Am J Physiol Regul Integr Comp Physiol. 2001;281(2):R417–26.
Lefrere I, De Coppet P, Camelin JC, Le Lay S, Mercier N, Elshourbagy N, et al. Neuropeptide AF and FF modulation of adipocyte metabolism. Primary insights from functional genomics and effects on beta-adrenergic responsiveness. J Biol Chem. 2002;277:39169–78.
Alfalah M, Michel MC. Neuropeptide Y and related peptides. Springer; 2004.
Chandrasekharan B, Nezami BG, Srinivasan S. Emerging neuropeptide targets in inflammation: NPY and VIP. Am J Physiol Gastrointest Liver Physiol. 2013;304:G949–57.
Sohn JW, Elmquist JK, Williams KW. Neuronal circuits that regulate feeding behavior and metabolism. Trends Neurosci. 2013;36:504–12.
Zhu H, Perkins C, Mingler MK, Finkelman FD, Rothenberg ME. The role of neuropeptide S and neuropeptide S receptor 1 in regulation of respiratory function in mice. Peptides. 2011;32:818–25.
Gajjar S, Patel BM. Neuromedin: an insight into its types, receptors and therapeutic opportunities. Pharmacol Rep. 2017;69:438–47.
Robinson SL, Thiele TE. A role for the neuropeptide somatostatin in the neurobiology of behaviors associated with substances abuse and affective disorders. Neuropharmacology. 2020;167: 107983.
Wojciechowski P, Andrzejewski K, Kaczynska K. Intracerebroventricular neuropeptide FF diminishes the number of apneas and cardiovascular effects produced by opioid receptors’ activation. Int J Mol Sci. 2020;21:8931.
Li F, Jiang H, Shen X, Yang W, Guo C, Wang Z, et al. Sneezing reflex is mediated by a peptidergic pathway from nose to brainstem. Cell. 2021;184:3762-73.e10.
Vazquez JM, Sulak M, Chigurupati S, Lynch VJ. A zombie LIF gene in elephants is upregulated by TP53 to induce apoptosis in response to DNA damage. Cell Rep. 2018;24:1765–76.
Acknowledgements
We acknowledge the various genome consortiums for sequencing and assembling the genomes.
Funding
This work is a result of the projects ATLANTIDA (Grant No. NORTE-01–0145-FEDER-000040), supported by the Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement and through the European Regional Development Fund (ERDF), and Marma-Detox (project no. 334739) funded by the Research Council of Norway. One PhD fellowship for author RV (SFRH/BD/144786/2019) was granted by Fundação para a Ciência e Tecnologia (FCT, Portugal) under the auspices of Programa Operacional Regional Norte (PORN), supported by the European Social Fund (ESF) and Portuguese funds (MECTES). FA had the support of FCT throughout the strategic projects UIDB/04292/2020 granted to MARE and LA/P/0069/2020 granted to the Associate Laboratory ARNET.
Author information
Authors and Affiliations
Contributions
RV, MC, RR and LFCC conceived the project and designed the research. RV, BP and AM performed the research. RV, MC, FA, ISP, RR and LFCC analysed data. RV and MC wrote the manuscript with input from all authors. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12915_2024_1984_MOESM1_ESM.xls
Additional file 1: Additional tables. Table S1 Screened mammalian species in the present study, including Cetacea. This includes, for each species, the corresponding common name, order, inspected genome assembly and database. Table S2 PseudoIndex value calculated for NPFF in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S3 Identified NPFF-disrupting mutations per mammal species and exon. Table S4 PseudoIndex value calculated for NPFFR1 in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S5 PseudoIndex value calculated for NPFFR2 in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S6 Identified NPFFR1-disrupting mutations per mammal species and exon. Table S7 Identified NPFFR2-disrupting mutations per mammal species and exon. Table S8 PseudoIndex value calculated for QRFP in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S9 Identified QRFP-disrupting mutations per mammal species and exon. Table S10 PseudoIndex value calculated for QRFPR in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S11 Identified QRFPR-disrupting mutations per mammal species and exon. Table S12 PseudoIndex value calculated for NPS in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S13 Identified NPS-disrupting mutations per mammal species and exon. Table S14 PseudoIndex value calculated for NPSR1 in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S15 Identified NPSR1-disrupting mutations per mammal species and exon. Table S16 PseudoIndex value calculated for NPB in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S17 Identified NPB-disrupting mutations per mammal species and exon. Identified QRFPR-disrupting mutations per mammal species and exon. Table S18 PseudoIndex value calculated for NPBWR2 in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S19 Identified NPBWR2-disrupting mutations per mammal species and exon. Table S20 PseudoIndex value calculated for NMBR in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S21 Identified NMBR-disrupting mutations per mammal species and exon. Table S22 PseudoIndex value calculated for SSTR4 in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S23 Identified SSTR4-disrupting mutations per mammal species and exon. Table S24 PseudoIndex value calculated for Npy6r in selected mammalian species. The table also includes the ID of the genomic sequence from where each target species’ input sequence was extracted, further imported into the analysis. Species displaying a PseudoIndex > 2 are highlighted in red. Table S25 Identified Npy6r-disrupting mutations per mammal species and exon. Table S26 In-depth description of the transcriptomic NCBI Sequence Read Archiveprojects, scrutinized in the transcriptomic analysis of the 4 represented cetaceans. Table S27 Gene expression in different tissues for four different cetacean species. Table S28 Transcriptomic read count of NMBR, NPB, NPFF, NPFFR1, NPFFR2, NPS, NPSR1 and QRFPR in Balaena mysticetus, Balaenoptera acutorostrata, Delphinapterus leucasand Monodon monoceros. Table S29 Tests for selection relaxation on the Mysticeti branch for NMBR, NPSR1, NPB, QRFP and NPS. Log-likelihood values and parameter estimates. Table S30 Tests for positive selection on the Mysticeti branch for NMBR. Log-likelihood values and parameter estimates. Table S31 Sequences of non-cetacean mammals incorporated in selection analysis. These include previously annotated genomic sequences at NCBI.
12915_2024_1984_MOESM2_ESM.pdf
Additional file 2: SRA validation of NPFF inactivating mutations in mammals. Fig. S1 SRA validation of absence of start codon in exon 1 of NPFF in Balaenoptera acutorostrata scammoni. Fig. S2 SRA validation of 14 nucleotide insertion in exon 2 of NPFF in Eschrichtius robustus. Fig. S3 SRA validation of 1 nucleotide deletion in exon 2 of NPFF in Monodon monoceros. Fig. S4 SRA validation of 1 nucleotide insertion and an in-frame premature stop codon in exon 2 of NPFF in Pteropus vampyrus.
12915_2024_1984_MOESM3_ESM.pdf
Additional file 3: SRA validation of NPFFR1 inactivating mutations in mammals. Fig. S1 SRA validation of 11 nucleotide deletion in exon 4 of NPFFR1 in Tursiops aduncus. Fig. S2 SRA validation of an in-frame premature stop codon in exon 4 of NPFFR1 in Artibeus jamaicensis. Fig. S3 SRA validation of 1 nucleotide insertion in exon 4 of NPFFR1 in Phascolarctos cinereus. Fig. S4 SRA validation of 1 nucleotide insertion in exon 4 of NPFFR1 in Manis javanica. Fig. S5 SRA validation of 2 nucleotide insertion in exon 4 of NPFFR1 in Equus asinus. Fig. S6 SRA validation of 1 nucleotide deletion in exon 4 of NPFFR1 in Marmota marmota marmota. Fig. S7 SRA validation of 2 nucleotide deletion in exon 4 of NPFFR1 in Marmota marmota marmota. Fig. S8 SRA validation of 5 nucleotide deletion in exon 4 of NPFFR1 in Camelus dromedarius. Fig. S9 SRA validation of 5 nucleotide deletion in exon 4 of NPFFR1 in Camelus dromedarius. Fig. S10 SRA validation of 4 nucleotide deletion in exon 4 of NPFFR1 in Camelus dromedarius.
12915_2024_1984_MOESM4_ESM.pdf
Additional file 4: SRA validation of NPFFR2 inactivating mutations in mammals. Fig. S1 SRA validation of an in-frame premature stop codon in exon 3 of NPFFR2 in Balaenoptera acutorostrata scammoni. Fig. S2 SRA validation of an in-frame premature stop codon in exon 3 of NPFFR2 in Mesoplodon bidens. Fig. S3 SRA validation of an in-frame premature stop codon in exon 3 of NPFFR2 in Mesoplodon densirostris. Fig. S4 SRA validation of an in-frame premature stop codon in exon 3 of NPFFR2 in Heterocephalus glaber. Fig. S5 SRA validation of 2 nucleotide insertion in exon 3 of NPFFR2 in Eschrichtius robustus. Fig. S6 SRA validation of 1 nucleotide deletion in exon 3 of NPFFR2 in Eubalaena japonica. Fig. S7 SRA validation of 2 nucleotide deletion in exon 3 of NPFFR2 in Globicephala melas. Fig. S8 SRA validation of 2 nucleotide deletion in exon 3 of NPFFR2 in Physeter macrocephalus. Fig. S9 SRA validation of 1 nucleotide insertion in exon 3 of NPFFR2 in Camelus ferus. Fig. S10 SRA validation of 1 nucleotide deletion in exon 1 of NPFFR2 in Canis lupus familiaris. Fig. S11 SRA validation of 1 nucleotide deletion in exon 1 of NPFFR2 in Vulpes vulpes.
12915_2024_1984_MOESM5_ESM.pdf
Additional file 5: SRA validation of QRFP inactivating mutations in mammals. Fig. S1 SRA validation of 1 nucleotide insertion in exon 1 of QRFP in Balaenoptera musculus. Fig. S2 SRA validation of 1 nucleotide insertion in exon 1 of QRFP in Sarcophilus harrisii. Fig. S3 SRA validation of 1 nucleotide insertion in exon 1 of QRFP in Propithecus coquereli. Fig. S4 SRA validation of 1 nucleotide insertion in exon 1 of QRFP in Mus pahari. Fig. S5 SRA validation of 11 nucleotide deletion in exon 1 of QRFP in Balaenoptera physalus. Fig. S6 SRA validation of absence of start codon in exon 1 of QRFP in Lagenorhynchus obliquidens. Fig. S7 SRA validation of 2 nucleotide insertion in exon 1 of QRFP in Mesoplodon densirostris. Fig. S8 SRA validation of 8 nucleotide deletion in exon 1 of QRFP in Physeter macrocephalus. Fig. S9 SRA validation of 10 nucleotide deletion in exon 1 of QRFP in Platanista minor. Fig. S10 SRA validation of an in-frame premature stop codon in exon 1 of QRFP in Tursiops truncatus. Fig. S11 SRA validation of 1 nucleotide deletion in exon 1 of QRFP in Ziphius cavirostris. Fig. S12 SRA validation of 1 nucleotide deletion in exon 1 of QRFP in Pteropus vampyrus. Fig. S13 SRA validation of 1 nucleotide deletion in exon 1 of QRFP in Oryctolagus cuniculus. Fig. S14 SRA validation of 1 nucleotide deletion in exon 1 of QRFP in Microcebus murinus. Fig. S15 SRA validation of 1 nucleotide insertion and an in-frame premature stop codon in exon 1 of QRFP in Tachyglossus aculeatus.
12915_2024_1984_MOESM6_ESM.pdf
Additional file 6: SRA validation of QRFPR inactivating mutations in mammals. Fig. S1 SRA validation of an in-frame premature stop codon in exon 5 of QRFPR in Physeter macrocephalus. Fig. S2 SRA validation of an in-frame premature stop codon in exon 1 of QRFPR in Delphinapterus leucas. Fig. S3 SRA validation of an in-frame premature stop codon in exon 1 of QRFPR in Globicephala melas. Fig. S4 SRA validation of an in-frame premature stop codon in exon 1 of QRFPR in Zalophus californianus. Fig. S5 SRA validation of a loss of canonical splicing site in exon 3 of QRFPR in Monodon monoceros. Fig. S6 SRA validation of 2 nucleotide insertion in exon 2 of QRFPR in Eumetopias jubatus. Fig. S7 SRA validation of an in-frame premature stop codon in exon 6 of QRFPR in Artibeus jamaicensis. Fig. S8 SRA validation of an in-frame premature stop codon in exon 6 of QRFPR in Myotis myotis. Fig. S9 SRA validation of an in-frame premature stop codon in exon 6 of QRFPR in Dromiciops gliroides. Fig. S10 SRA validation of 8 nucleotide deletion and 2 nucleotide insertion in exon 4 of QRFPR in Pteropus vampyrus. Fig. S11 SRA validation of an in-frame premature stop codon and 2 nucleotide deletion in exon 6 of QRFPR in Vombatus ursinus. Fig. S12 SRA validation of 2 nucleotide deletion in exon 4 of QRFPR in Tachyglossus aculeatus.
12915_2024_1984_MOESM7_ESM.pdf
Additional file 7: SRA validation of NPS inactivating mutations in mammals. Fig. S1 SRA validation of 2 nucleotide insertion in exon 3 of NPS in Globicephala melas. Fig. S2 SRA validation of 2 nucleotide insertion in exon 3 of NPS in Lagenorhynchus acutus. Fig. S3 SRA validation of 4 nucleotide deletion in exon 3 of NPS in Camelus bactrianus. Fig. S4 SRA validation of an in-frame premature stop codon in exon 3 of NPS in Heterocephalus glaber. Fig. S5 SRA validation of an in-frame premature stop codon in exon 2 of NPS in Trichechus manatus latirostris.
12915_2024_1984_MOESM8_ESM.pdf
Additional file 8: SRA validation of NPSR1 inactivating mutations in mammals. Fig. S1 SRA validation of an in-frame premature stop codon in exon 7 of NPSR1 in Balaenoptera acutorostrata scammoni. Fig. S2 SRA validation of an in-frame premature stop codon in exon 7 of NPSR1 in Theropithecus gelada. Fig. S3 SRA validation of an in-frame premature stop codon in exon 5 of NPSR1 in Delphinapterus leucas. Fig. S4 SRA validation of an in-frame premature stop codon in exon 5 of NPSR1 in Physeter macrocephalus. Fig. S5 SRA validation of an in-frame premature stop codon in exon 5 of NPSR1 in Enhydra lutris kenyoni. Fig. S6 SRA validation of 1 nucleotide insertion in exon 5 of NPSR1 in Camelus ferus. Fig. S7 SRA validation of 1 nucleotide deletion in exon 1 of NPSR1 in Sus scrofa. Fig. S8 SRA validation of an in-frame premature stop codon in exon 6 of NPSR1 in Callorhinus ursinus. Fig. S9 SRA validation of an in-frame premature stop codon in exon 8 of NPSR1 in Phoca vitulina. Fig. S10 SRA validation of 2 nucleotide deletion in exon 1 of NPSR1 in Talpa occidentalis. Fig. S11 SRA validation of an in-frame premature stop codon in exon 3 of NPSR1 in Platanista minor. Fig. S12 SRA validation of an in-frame premature stop codon in exon 3 of NPSR1 in Fukomys damarensis. Fig. S13 SRA validation of 1 nucleotide deletion in exon 5 of NPSR1 in Peromyscus leucopus. Fig. S14 SRA validation of an in-frame premature stop codon in exon 2 of NPSR1 in Lontra canadensis.
12915_2024_1984_MOESM9_ESM.pdf
Additional file 9: SRA validation of NPB inactivating mutations in mammals. Fig. S1 SRA validation of 1 nucleotide insertion in exon 1 of NPB in Delphinapterus leucas. Fig. S2 SRA validation of 1 nucleotide insertion in exon 1 of NPB in Kogia breviceps. Fig. S3 SRA validation of 11 nucleotide deletion in exon 1 of NPB in Halichoerus grypus. Fig. S4 SRA validation of 5 nucleotide insertion in exon 1 of NPB in Rhinolophus ferrumequinum. Fig. S5 SRA validation of 5 nucleotide insertion in exon 1 of NPB in Trichechus manatus latirostris. Fig. S6 SRA validation of 4 nucleotide insertion in exon 2 of NPB in Bison bison bison. Fig. S7 SRA validation of absence of start codon in exon 1 of NPB in Camelus bactrianus.
12915_2024_1984_MOESM10_ESM.pdf
Additional file 10: SRA validation of NPBWR2 inactivating mutations in mammals. Fig. S1 SRA validation of an in-frame premature stop codon in exon 1 of NPBWR2 in Delphinapterus leucas. Fig. S2 SRA validation of an in-frame premature stop codon in exon 1 of NPBWR2 in Otolemur garnettii. Fig. S3 SRA validation of an in-frame premature stop codon in exon 1 of NPBWR2 in Loxodonta africana. Fig. S4 SRA validation of an in-frame premature stop codon in exon 1 of NPBWR2 in Meriones unguiculatus. Fig. S5 SRA validation of 11 nucleotide deletion in exon 1 of NPBWR2 in Physeter macrocephalus. Fig. S6 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Ailuropoda melanoleuca. Fig. S7 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Halichoerus grypus. Fig. S8 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Mustela erminea. Fig. S9 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Panthera pardus. Fig. S10 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Suricata suricatta. Fig. S11 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Talpa occidentalis. Fig. S12 SRA validation of 1 nucleotide deletion in exon 1 of NPBWR2 in Ceratotherium simum simum. Fig. S13 SRA validation of 14 nucleotide deletion in exon 1 of NPBWR2 in Ochotona princeps. Fig. S14 SRA validation of 2 nucleotide deletion in exon 1 of NPBWR2 in Jaculus jaculus. Fig. S15 SRA validation of 1 nucleotide insertion in exon 1 of NPBWR2 in Rhinopithecus roxellana.
12915_2024_1984_MOESM11_ESM.pdf
Additional file 11: SRA validation of NMBR inactivating mutations in mammals. Fig. S1 SRA validation of 1 nucleotide insertion in exon 2 of NMBR in Delphinapterus leucas. Fig. S2 SRA validation of 1 nucleotide insertion in exon 2 of NMBR in Mesocricetus auratus. Fig. S3 SRA validation of 1 nucleotide deletion in exon 2 of NMBR in Globicephala melas. Fig. S4 SRA validation of 1 nucleotide deletion in exon 2 of NMBR in Rhinopithecus roxellana. Fig. S5 SRA validation of 11 nucleotide deletion and an in-frame premature stop codon in exon 3 of NMBR in Kogia breviceps. Fig. S6 SRA validation of 7 nucleotide deletion in exon 1 of NMBR in Ziphius cavirostris. Fig. S7 SRA validation of 7 nucleotide deletion in exon 1 of NMBR in Physeter macrocephalus. Fig. S8 SRA validation of 1 nucleotide insertion in exon 1 of NMBR in Cavia porcellus.
12915_2024_1984_MOESM12_ESM.pdf
Additional file 12: SRA validation of SSTR4 inactivating mutations in mammals. Fig. S1 SRA validation of 2 nucleotide deletion in exon 1 of SSTR4 in Tursiops truncatus. Fig. S2 SRA validation of 2 nucleotide deletion in exon 1 of SSTR4 in Fukomys damarensis. Fig. S3 SRA validation of 2 nucleotide deletion in exon 1 of SSTR4 in Heterocephalus glaber. Fig. S4 SRA validation of 2 nucleotide deletion in exon 1 of SSTR4 in Orycteropus afer afer. Fig. S5 SRA validation of 1 nucleotide deletion in exon 1 of SSTR4 in Hippopotamus amphibius. Fig. S6 SRA validation of 1 nucleotide deletion in exon 1 of SSTR4 in Phascolarctos cinereus. Fig. S7 SRA validation of 1 nucleotide deletion in exon 1 of SSTR4 in Leptonychotes weddellii. Fig. S8 SRA validation of 1 nucleotide deletion in exon 1 of SSTR4 in Manis javanica. Fig. S9 SRA validation of 1 nucleotide deletion in exon 1 of SSTR4 in Peromyscus leucopus. Fig. S10 SRA validation of 1 nucleotide deletion in exon 1 of SSTR4 in Trichechus manatus latirostris. Fig. S11 SRA validation of 5 nucleotide deletion in exon 1 of SSTR4 in Chrysochloris asiatica. Fig. S12 SRA validation of 1 nucleotide insertion and an in-frame premature stop codon in exon 1 of SSTR4 in Callorhinus ursinus. Fig. S13 SRA validation of 47 nucleotide deletion in exon 1 of SSTR4 in Vulpes vulpes. Fig. S14 SRA validation of an in-frame premature stop codon in exon 1 of SSTR4 in Hipposideros armiger. Fig. S15 SRA validation of an in-frame premature stop codon in exon 1 of SSTR4 in Sorex araneus. Fig. S16 SRA validation of an in-frame premature stop codon in exon 1 of SSTR4 in Equus asinus. Fig. S17 SRA validation of 5 nucleotide insertion in exon 1 of SSTR4 in Rhinolophus ferrumequinum. Fig. S18 SRA validation of 1 nucleotide insertion and 8 nucleotide deletion in exon 1 of SSTR4 in Galeopterus variegatus. Fig. S19 SRA validation of 54 nucleotide insertion in exon 1 of SSTR4 in Tachyglossus aculeatus. Fig. S20 SRA validation of 2 nucleotide insertion in exon 1 of SSTR4 in Loxodonta africana.
12915_2024_1984_MOESM13_ESM.pdf
Additional file 13: SRA validation of Npy6r inactivating mutations in mammals. Fig. S1 SRA validation of 1 nucleotide insertion in exon 1 of Npy6r in Tursiops truncatus. Fig. S2 SRA validation of 1 nucleotide insertion in exon 1 of Npy6r in Dasypus novemcinctus. Fig. S3 SRA validation of 1 nucleotide insertion in exon 1 of Npy6r in Nomascus leucogenys. Fig. S4 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Bos taurus. Fig. S5 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Felis catus. Fig. S6 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Choloepus didactylus. Fig. S7 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Talpa occidentalis. Fig. S8 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Callithrix jacchus. Fig. S9 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Carlito syrichta. Fig. S10 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Cercocebus atys. Fig. S11 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Otolemur garnettii. Fig. S12 SRA validation of an in-frame premature stop codon in exon 1 of Npy6r in Rhinopithecus bieti. Fig. S13 SRA validation of a 1 nucleotide insertion and an in-frame premature stop codon in exon 1 of Npy6r in Ursus maritimus. Fig. S14 SRA validation of 2 nucleotide deletion in exon 1 of Npy6r in Rhinolophus ferrumequinum. Fig. S15 SRA validation of 4 nucleotide deletion in exon 1 of Npy6r in Ochotona princeps. Fig. S16 SRA validation of 4 nucleotide deletion in exon 1 of Npy6r in Pan paniscus. Fig. S17 SRA validation of a 1 nucleotide deletion and an in-frame premature stop codon in exon 1 of Npy6r in Sapajus apella. Fig. S18 SRA validation of a 1 nucleotide deletion and an in-frame premature stop codon in exon 1 of Npy6r in Rattus rattus. Fig. S19 SRA validation of 2 nucleotide deletion in exon 1 of Npy6r in Trachypithecus francoisi. Fig. S20 SRA validation of 2 nucleotide deletion in exon 1 of Npy6r in Galeopterus variegatus. Fig. S21 SRA validation of 8 nucleotide insertion and 1 nucleotide deletion in exon 1 of Npy6r in Cavia porcellus. Fig. S22 SRA validation of 14 nucleotide deletion in exon 1 of Npy6r in Loxodonta africana.
12915_2024_1984_MOESM14_ESM.pdf
Additional file 14: SRA validation of inactivating mutations in NPFF transcripts of cetaceans. Fig. S1 Presence of a premature stop codon in exon 3 of the corresponding species NPFF transcripts in Balaena mysticetus. Fig. S2 Presence of a premature stop codon in exon 3 of the corresponding species NPFF transcripts in Monodon monoceros.
12915_2024_1984_MOESM15_ESM.pdf
Additional file 15:SRA validation of inactivating mutations in NPFFR1 transcripts of cetaceans. Fig. S1 Lack of terminal stop codon in exon 4 of the corresponding species NPFFR1 transcripts in Delphinapterus leucas. Fig. S2 Lack of terminal stop codon in exon 4 of the corresponding species NPFFR1 transcripts in Monodon monoceros.
12915_2024_1984_MOESM16_ESM.pdf
Additional file 16: SRA validation of inactivating mutations in NPFFR2 transcripts of cetaceans. Fig. S1 Presence of a premature stop codon in exon 3 of the corresponding species NPFFR2 transcripts in Balaena mysticetus. Fig. S2 Presence of a premature stop codon in exon 3 of the corresponding species NPFFR2 transcripts in Delphinapterus leucas. Fig. S3 Presence of a premature stop codon in exon 1 of the corresponding species NPFFR2 transcripts in Monodon monoceros.
12915_2024_1984_MOESM17_ESM.pdf
Additional file 17: SRA validation of inactivating mutations in QRFPR transcripts of cetaceans. Fig. S1 Presence of a premature stop codon in exon 5 of the corresponding species QRFPR transcripts in Balaena mysticetus. Fig. S2 Presence of a premature stop codon in exon 5 of the corresponding species QRFPR transcripts in Balaenoptera acutorostrata scammoni. Fig. S3 Presence of a premature stop codon in exon 1 of the corresponding species QRFPR transcripts in Delphinapterus leucas.
12915_2024_1984_MOESM18_ESM.pdf
Additional file 18: SRA validation of inactivating mutations in NPSR1 transcripts of cetaceans. Fig. S1 Presence of a premature stop codon in exon 3 of the corresponding species NPSR1 transcripts in Delphinapterus leucas.
12915_2024_1984_MOESM19_ESM.pdf
Additional file 19: SRA validation of inactivating mutations in NPB transcripts of cetaceans. Fig. S1 Lack of terminal stop codon in exon 2 of the corresponding species NPB transcripts in Delphinapterus leucas. Fig. S2 Lack of terminal stop codon in exon 2 of the corresponding species NPB transcripts in Monodon monoceros.
12915_2024_1984_MOESM20_ESM.pdf
Additional file 20: SRA validation of inactivating mutations in NMBR transcripts of cetaceans. Fig. S1 Presence of a premature stop codon in exon 1 of the corresponding species NMBR transcripts in Delphinapterus leucas. Fig. S2 Presence of a premature stop codon in exon 2 of the corresponding species NMBR transcripts in Monodon monoceros.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Valente, R., Cordeiro, M., Pinto, B. et al. Alterations of pleiotropic neuropeptide-receptor gene couples in Cetacea. BMC Biol 22, 186 (2024). https://doi.org/10.1186/s12915-024-01984-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-024-01984-0