
Abstract
Background
Lung cancer is a high-incidence cancer, and it is also the most common cause of cancer death worldwide. 80–85% of lung cancer cases can be classified as non-small cell lung cancer (NSCLC).
Methods
NSCLC transcriptome data and clinical information were downloaded from the TCGA database and GEO database. Firstly, we analyzed and identified the differentially expressed genes (DEGs) between non-metastasis group and metastasis group of NSCLC in the TCGA database, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) were consulted to explore the functions of the DEGs. Thereafter, univariate Cox regression and LASSO Cox regression algorithms were applied to identify prognostic metastasis-related signature, followed by the construction of the risk score model and nomogram for predicting the survival of NSCLC patients. GSEA analyzed that differentially expressed gene-related signaling pathways in the high-risk group and the low-risk group. The survival of NSCLC patients was analyzed by the Kaplan–Meier method. ROC curve was plotted to evaluate the accuracy of the model. Finally, the GEO database was further applied to verify the metastasis‑related prognostic signature.
Results
In total, 2058 DEGs were identified. GO functions and KEGG pathways analysis results showed that the DEGs mainly concentrated in epidermis development, skin development, and the pathway of Neuro active ligand -receptor interaction in cancer. A six-gene metastasis-related risk signature including C1QL2, FLNC, LUZP2, PRSS3, SPIC, and GRAMD1B was constructed to predict the overall survival of NSCLC patients. The reliability of the gene signature was verified in GSE13213. The NSCLC patients were grouped into low-risk and high-risk groups based on the median value of risk scores. And low-risk patients had lower risk scores and longer survival time. Univariate and multivariate Cox regression verified that this signature was an independent risk factor for NSCLC.
Conclusion
Our study identified 6 metastasis biomarkers in the NSCLC. The biomarkers may contribute to individual risk estimation, survival prognosis.
Similar content being viewed by others

Introduction
Lung cancer is the leading cause of cancer-related death throughout the world. According to the World Health Organization (WHO), 2.2 million new lung cancer cases and 1.8 million fatalities are expected in 2020 [1]. NSCLC is the most common kind of lung cancer, accounting for 85 percent of all cases. Lung cancer patients die from invasiveness and metastasis in over 90% of cases, resulting in a 5-year survival rate of barely 15% [2]. As a result, research into the major regulators of metastasis is crucial for improving lung cancer treatment.
Because 80–85 percent of patients are first identified with either unresectable or metastatic tumors, despite breakthroughs in diagnostic tools, radiotherapies, and systemic treatments for NSCLC, the five-year overall survival (OS) remains at 10% [3,4,5]. The five-year survival rate post-operation was 20% for the tiny number of patients with a resectable and confined malignancy [3]. The prognosis of individuals with NSCLC is commonly believed to be determined by metastasis [2, 6]. Patients with metastasis have a shorter survival rate than those with localized malignancies, with just 6–8 months on average [7, 8]. The brain is the most common distant metastatic site for NSCLC [6, 9]. As a result, it's important to investigate possible biomarkers that might differentiate individuals with a poor prognosis based on tumor metastasis-related genes.
The most likely site of lung cancer metastasis is the brain, the development of the modified extracellular matrix (ECM), angiogenesis for micro-metastatic, and the building of immune escape are all part of the NSCLC metastasis process[10]. Lung cancers are exceedingly varied at both the cellular and molecular levels, according to previous research [11, 12]. Molecular markers are becoming more important in predicting the prognosis of individuals with NSCLC [12, 13]. Many prognostic models with excellent predictive value have been constructed using public resources such as The Cancer Genome Atlas (TCGA), International Cancer Genome Consortium (ICGC), and Gene Expression Omnibus (GEO) [14,15,16]. By mining the TCGA data, Dong et al. recently demonstrated that the Liver-Metastasis-Related Genes have high predictive potential for predicting the clinical outcomes of patients with pancreatic adenocarcinoma [17]. However, limited study on mRNA combination biomarkers for NSCLC metastasis has been done. We predicted in this study that differentially expressed genes linked to metastasis could be able to predict the prognosis of NSCLC patients. The mRNA expression data of NSCLC tissues in M0 stage and M1 stage from the TCGA datasets were combined in this study. Following cox and lasso regression, a six-gene prognostic signature was created with the potential to predict survival time for NSCLC patients.
Materials and methods
Data source
The transcriptome and clinical data were downloaded from the TCGA database (https://portal.gdc.cancer.gov/), including metastasis samples (n = 31) and non-metastatic samples (n = 733), and were used as training set. 117 LUAD samples from GEO datasets, the accession number of GEO datasets is GSE13213, and were used as external validation sets.
Identification of DEGs
The ‘Limma’ package [18] in the R statistical software was used to identify DEGs between the metastatic group and the non-metastatic group, with adj p value < 0.05 set as the screening thresholds. A heat map cluster and volcano plot of the DEGs were created using the “pheatmap” and “ggplots” packages via R software.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) analysis
To explore the potential functions of the metastasis-related gene signature, GO analysis and KEGG enrichment analysis were conducted by the ‘clusterProfiler’ [19] package. P.adjust < 0.05 were found to be statistically relevant.
Univariate cox regression and lasso regression analysis
We first used the R package survival coxph function to perform Univariate Cox Regression analysis on DEGs to screen metastasis-related genes significantly related to the survival. p < 0.05 was selected as the threshold for filtering. Moreover, the screened prognosis-related metastasis-related genes were incorporated into Lasso regression model, in which penalties were applied to above gene for preventing overfitting effects of the model. We performed LASSO Cox Regression analysis and identified 12 signature genes [24]. At last, multivariate COX regression analysis constructed the prognostic model successfully. The patients of train and validation sets were divided into low- and high-risk groups on the foundation of the median value of the risk score of train cohort, respectively. Survival differences between the two groups were assessed by Kaplan–Meier. Meanwhile, univariate and multivariate prognostic analyses (p < 0.05) were performed for the training group to determine whether the riskScore obtained from the model could be an independent prognostic factor.
Drawing and validation of the nomogram
A nomogram was established with the independent risk factors such as clinical information and risk score to predict the possibility of 1-year, 3-year and 5-year OS of NSCLC patients. The efficacy of the nomogram was evaluated by the calibration curve.
Estimation of immune score, stromal score, and tumor purityimmune infiltration
The ESTIMATE package was used to calculate the immune score (representing the level of immune cell infiltration) and stromal score (representing the number of stroma) of each PAAD sample. The ESTIMATE score was defined as the sum of the immune and stromal scores. Then, the differences in stromal score, immune score, ESTIMATE scores, tumor purity scores between high-risk groups and low-risk groups were compared by the Wilcoxon test. p value < 0.05 was considered significant [16]. To predict the effect of immune checkpoint blockade therapy, we also explored the expression of immune checkpoint genes in the groups.
Estimation of relationship between this prognostic risk model and clinical characteristics and tumor mutation burden (TMB)
We evaluated the relationship between the Risk score and clinical characteristics acquired from TCGA, as follows: M (M0 and M1), N (N0 and N1-3), T (T1-2 and T3-4), and stage (I-II and III-IV). The tumor mutational data of NSCLC patients were obtained from TCGA database, and tumor mutational burden (TMB) was calculated for each NSCLC patient.
The analysis of GSEA
The R package “limma” was used to analyze differential expression between high-risk and low-risk groups [19], and all genes were ranked by fold change values. h.all.v7.4.symbols.gmt data set is downloaded from MSigDB, Gene Set Enrichment Analysis was performed to clarify the significant annotated pathways through R package “clusterProfiler".
Results
Identification of DEGs
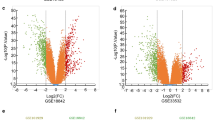
The 764 NSCLC samples in the TCGA dataset were separated into two groups: non-metastatic (31 samples) and metastatic (733 samples). The TCGA dataset yielded 2058 DEGs (Fig. 1A–B), 1499 of which were down-regulated and 559 of which were up-regulated.

Identification of differential expressed gene between non-metastatic and metastatic group in NSCLC. A The volcano plot demonstrating the differentially expressed genes. B Heat map of differentially expressed genes in NSCLC
Functional enrichment analysis
DEGs' biological functions and pathways can be studied via gene enrichment analysis. Epidermis development, skin development, epidermal cell differentiation, keratinocyte differentiation, and keratinization are among the biological processes enriched in GO (top 5). Presynapse, synaptic membrane, glutamatergic synapse, and intermediate filament intermediate filament are the biological components of GO (top 5). Peptidase regulator activity, endopeptidase regulator activity, endopeptidase inhibitor activity, peptidase inhibitor activity, and serine-type endopeptidase inhibitor activity are the top five molecular functions of GO (Fig. 2A). Similarly, neuroactive ligand-receptor interaction, chemical carcinogenesis-receptor activation, estrogen signaling route, staphylococcus aureus infection, and drug metabolism-cytochrome P450 are the top five significantly enriched pathways (Fig. 2B).

Representative results of GO and KEGG analyses. A The molecular functions of the 6 screened genes. B The potential biological pathways of the screened genes. Data from KEGG website (KEGG: Kyoto Encyclopedia of Genes and Genomes)
Construction and validation of the risk score model based on 6 prognostic metastasis-related genes
The DEGs in the TCGA training group were subjected to univariate Cox regression analysis. The findings of the univariate regression analysis revealed that genes related to metastasis were substantially correlated with NSCLC patients' prognosis (p 0.05). (Fig. 3A). For these genes having prognostic value, LASSO regression analysis was used to avoid over-fitting the prognostic model. The LASSO regression analysis revealed that 12 genes had a significant relationship with OS (Fig. 3B and C). Finally, we ran a multivariate regression analysis on the 12 genes we chose. C1QL2, FLNC, LUZP2, PRSS3, SPIC, and GRAMD1B were identified as risk variables for OS in the TCGA training group by multivariate regression analysis (Fig. 3D). The risk score was calculated as (− 0.265 × C1QL2) + (0.227 × FLNC) + (− 0.625 × LUZP2) + (0.095 × PRSS3) + (0.193 × SPIC) + (0.447 × GRAMD1B). Following that, the TCGA patients were split into high- and low-risk groups based on the median risk scores. Patients with high-risk scores had worse survival rates in the training set by the Kaplan–Meier curves (p 0.0001). (Fig. 3E). Similarly, 117 individuals from GSE13213 were chosen as the validation cohort and classified into high- and low-risk groups based on the median risk score, with the same risk score calculation formula as the TCGA cohort. The survival curve revealed a significant difference (p 0.05) between the two groups (Fig. 3F). The relationship between the RiskScore and clinical features was analyzed, and it was found that the risk score constructed based on the six-gene signature distinguished the high- and low-risk groups according to age, M0 stage, N stage, stage I-II, T1-2 stage. This finding consequently indicated that the risk model had a strong predictive ability across clinical features.

Construction of risk signature in the TCGA cohort. A Univariate Cox analysis of differentially expressed genes. (B) Cross-validation for tuning the parameter selection in the LASSO regression. C LASSO regression of the differentially expressed genes. D Multivariate Cox analysis of differentially expressed genes. E–F K–M survival analysis of risk prognostic model of NSCLC patients in TCGA
The expression of RiskScore on different clinical features and the construction of nomogram
The multivariable Cox method was used to find the three independent prognostic indicators (age, stage, and risk score) of NSCLC patients in the TCGA data set (Fig. 4A). Following that, a nomogram for 1-year, 3-year, and 5-year survival rates was produced based on the age, stage, and risk score to objectively estimate the survival likelihood of each NSCLC patient (Fig. 4B). In addition, calibration curves for 1-year, 3-year, and 5-year survival rates were plotted to test the nomogram's accuracy, with the findings revealing that the nomogram-predicted and actual survival probability are generally in accord (Fig. 4C–E). Patients in the TCGA cohort were divided into high- and low-risk groups based on the median risk score computed from the nomogram. Figure 4F indicated that patients in the high-risk group had significantly shorter OS than those of the low-risk group (p < 0.001).

Construction and evaluation of prognostic models based on risk scores and clinical features. A Forest plot for multivariate COX regression analysis based on risk scores and clinical features. B A nomogram predicts the risk of progression in patients with NSCLC by four clinicopathological features. C–E The calibration curve is used to evaluate the accuracy of one-, three-, and five-year progress forecasts of nomograms. F K–M curves of prognostic models based on risk scores and clinical features
The correlation between the prognostic risk model and clinical pathological characteristics of patients
We started by looking at the association between risk scores and clinical variables. The results revealed that there was no significant difference in risk ratings among N stages (Fig. 5A). We looked at the differences in risk scores between different NSCLC groups. The subgroup analysis stratified by stage revealed that stage IV NSCLC patients had a significantly higher risk score than stage I NSCLC patients (p = 0.0031). (Fig. 5B). Furthermore, M1 NSCLC patients had a significantly higher risk score compared to M0 NSCLC patients (p = 0.043). In addition, T3NSCLC patients had a considerably higher risk score than T1 NSCLC patients (p = 0.0052). (Fig. 5C–D).

The correlation between the prognostic risk model and clinical pathological characteristics (stage, TNM) A–D
Analysis of the relationship between the immune microenvironment and risk score model in NSCLC patients
Using the ESTIMATE algorithm, we estimated the stromal score, immune score, and tumor purity of NSCLC from TCGA dataset. Our data showed that the immune score and stromal score of the high-risk group was significantly higher than those of the low-risk group (Fig. 6A), and the tumor purity score of the high-risk group was significantly lower than that of the low-risk group. To further explore the individual immune microenvironment and develop individualized treatment, immune infiltration and immune checkpoint genes in high- and low-risk group were further investigated (Fig. 6B–C). The low-risk group had considerably lower markers of Macrophages, Macrophages M1, MEP, Monocytes, pDC, and Th2 cells than the high-risk group. The low-risk group, on the other hand, showed increased Th1 cell, MEP, and HSC marker expression. In addition, variations in immune checkpoint genes were discovered in the high-risk and low-risk groups. TNFSF15 was expressed at higher levels in the low-risk group than in the high-risk group. When compared to the low-risk group, the high-risk group showed higher expressions of ADORA2A, TNFSF14, CD28, ICOS, TIGIF, TNFRSF9, CD276, TNFSF9, TNFRSF8, PDCD1, CTLA4, TNFSF4, CD86, NRP1, TNFRSF4, CD70, LAIR1, C10orf54, HAVCR2, and CD200.

Analysis of the Relationship Between the Immune Microenvironment and Risk Score Model in NSCLC Patients. A ESTIMATE-analysis of the high and low risk groups. B Analysis of the immune-infiltrating cells. CMolecular analysis of immune checkpoints in high and low risk groups. D TMB scores for the high and low risk group
We also estimated the TMB of each sample and discovered that in the TCGA dataset, TMB was significantly greater in the high-risk group (p = 0.0056). (Fig. 6D).
GSEA analysis
GSEA analyses were conducted to further explore the difference biological mechanism between low- and high-risk groups. we found that signaling pathway (Fig. 7), including allograft rejection, coagulation, complement, epithelial mesenchymal transition, G2M checkpoint, IL6-JAK-STAT3 signaling, inflammatory response, interferon gamma response, KRAS signaling up, TNFA signaling via NFkB were significantly enriched in the high-risk group.

Gene Set Enrichment Analysis. Differences in gene sets between high and low risk groups
Discussion
In this article, NSCLC samples were classified into metastatic group and non-metastatic group according to M stage. TCGA was used as training cohort and construct a prognostic model, while GEO database was used as validation cohort to verify the efficacy of the prognostic model evaluation. Firstly, we analyzed the gene expression data and clinical data of NSCLC patients enrolled in TCGA, discerning 2058 DEGs related to metastasis. Using univariate, LASSO and multivariate Cox regression analysis, 6 mRNAs (C1QL2, FLNC, LUZP2, PRSS3, SPIC, GRAMD1B) had been found as independent prognosis predictors in NSCLC. Secondly, survival analysis was utilized to examine the availability of the prognostic model. The expression pattern of all the 6 mRNAs, had a correlation to OS which meant that with the generate of these mRNAs’ expression, patients would have a different survival time. Thirdly, the model constructed in training group was validated externally, adding dependability to the outcomes.
Through pathway enrichment analysis of metastasis-related genes, we found that many GO pathways were enriched, such as epidermis development, skin development, epidermal cell differentiation, keratinocyte differentiation, and so on. Many of them have been confirmed to be associated with tumor metastasis. Close relationship, for example, Sabounsji's study pointed out that the metastasis of NSCLC is closely related to epidermal cell differentiation [20]. A correlation between keratinocyte differentiation and Metastatic Melanoma was also pointed out in Li’s studies [21]. The mRNAs in model had been reported in other articles that they also had relationship with different types of cancers. A study from Sigin et al. found that in in luminal B breast cancer the methylation level of C1QL2 is closely linked to neoadjuvant chemotherapy in luminal B breast cancer patients [22]. Filamin C (FLNC) is a large actin-cross-linking protein that is found in a variety of cells. According to the previous literature, temporary expression or silencing of FLNC can alter cancer cell proliferation and colony formation, whereas endogenous FLNC silencing can accelerate cancer cell motility and invasion [23]. LUZP2(leucine zipper protein 2 gene), located on Chr 11p13–11p14 and encoding a leucine zipper protein, has been shown to be deleted in Wilms' tumor patients. Wilms' tumor, genital abnormalities, aniridia, and mental retardation is a rare congenital abnormality syndrome characterized by Wilms' tumor, genital deformities, aniridia, and mental retardation [24, 25]. Furthermore, Zhao et colleagues found that LUZP2 mRNA expression is elevated in hormone-naive prostate cancer (PC) relative to normal prostate tissues, but downregulated throughout the progression from hormone-naive PC to castration-resistant PC (CRPC) [26]. PRSS3 (serine protease 3) is a member of the serine protease family that is produced in pancreatic acinar cells and released into the small intestine to help in digestion. According to Wang's findings, increased PRSS3 expression may enhance stomach cancer metastasis and serve as an independent molecular indication of poor patient prognosis [27]. SpiC is a member of Spi subtypes, SpiC has crucial functions in myeloid differentiation, however, there have been no reports of the role of SpiC in tumors[28]. GRAMD1B (GRAM domain-containing protein 1B) was identified as a putative component of the signaling cascade17, has been implicated in human malignancies [29]. Specifically, it was reported to play a role in chemoresistance of ovarian cancer patients, such that GRAMD1B inhibition led to an anti-tumor effect [30]. Khanna’s study has proved that GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signalling [29]. Those results had represented similar conclusions as this study.
Tumor metastasis is triggered by interactions between cancer cells and numerous stromal cell components of the tumor microenvironment, as well as by the accumulation of intrinsic changes in malignant cells [31, 32]. Inflammation and infiltration of tumor tissue by immune cells from the host, such as tumor-associated macrophages, myeloid-derived suppressor cells, and regulatory T cells, have been demonstrated to promote tumor development as well as invasion and metastasis [33, 34]. Our data showed that the immune score and stromal score of the high-risk group was significantly higher than that of the low-risk group. such as macrophages, macrophages M1, monocytes, pDC and Th2 cells immune infiltration was significantly higher than the low-risk group. This suggests that tumor metastasis-related genes also play a role in regulating tumor immunity. To explain more detailed immune cell infiltration in NSCLC, ssGSEA was used to find the low-risk group had higher marker expression of iDC, MSC, Th2 cells, Endothelial cells, Monocytes. These results are in line with the conclusions of previous studies [35, 36], indicating that our prognostic model can not only have a good predictive effect on the prognosis of patients with NSCLC. And it can respond to patient immune changes to some extent. This will be very important for immunotherapy with NSCLC patients. For example, in the future, patient response to immunotherapy can be predicted through prognostic models established in our study.
We wished to understand more genetically the possible mechanisms by which our model worked, GSEA was performed to do enrichment analysis of high and low risk groups separately, which could be found including allograft rejection, coagulation, complement, epithelial mesenchymal transition, G2M checkpoint, IL6 JAK STAT3 signaling, inflammatory response, interferon gamma response, KRAS signaling up, TNFA signaling via NFkB were significantly enriched in the high-risk group. These pathways have all been shown in previous studies to be directly or indirectly related to tumor metastasis. For example, EMT, an evolutionarily conserved developmental program, has been linked to carcinogenesis and imparts metastatic qualities to cancer cells by increasing mobility, invasion, and resistance to apoptotic stimuli. Furthermore, EMT-derived tumor cells have stem cell characteristics and are very resistant to treatment [37]. The cytokine interleukin-6 (IL6) and its downstream effector STAT3 form a major oncogenic pathway in breast cancer that has been hypothesized to be functionally linked to estrogen receptor (ER). Siersbak et al. found that IL6/STAT3 signaling promotes metastasis in ER + breast cancer that is not ER positive. A subset of ER enhancers is hijacked by STAT3 to produce a unique transcriptional pathway [38]. Some of the potential pathways we have identified have been reported to be associated with tumor metastasis, which validates our results, and our results find potential pathways that have not been explored to metastasis. This provides new perspectives for future studies of genes for tumor metastasis.
Finally, we developed a model and a biomarker for predicting the prognosis of NSCLC metastases by a series of bioinformation analyses. Patients in the low-risk category had a superior overall survival rate than those in the high-risk group, according to our findings which were confirmed in both the train and test cohorts. Our study opened a new avenue for the diagnostic and therapy of NSCLC. However, there were still exist some limitation in this research. Firstly, the data in TCGA may contain varying degrees of mistake, and the amount of data contained is limited, which may lead to inaccuracies. Second, the lack of in vivo and in vitro research will result in insufficient evidence. Last, There is still a flaw in our study that the TCGA database cannot provide paired samples. Therefore, we cannot longitudinally compare the situation of the same patient with different transfer times, and we will also include more cohorts in future studies to make up for this deficiency. It is also worth mentioning that our study is not based on all clinical features, including age, gender, etc., but a prognostic model constructed from only some accessible clinical features. Such as T and N staging and so on. Future studies need to incorporate more clinical features to achieve better model performance. As a result, further research and trials are needed to verify the model and biomarker to assure its robustness.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the TCGA repository. The Cancer Genome Atlas Program—National Cancer Institute. The validation set from GEO database, the accession number of GEO datasets is GSE13213. GEO Accession viewer (nih.gov).
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–94.
Counago F, Luna J, Guerrero LL, Vaquero B, Guillen-Sacoto MC, Gonzalez-Merino T, et al. Management of oligometastatic non-small cell lung cancer patients: current controversies and future directions. World J Clin Oncol. 2019;10(10):318–39.
Alexander M, Lin E, Cheng H. Leptomeningeal metastases in non-small cell lung cancer: optimal systemic management in NSCLC with and without driver mutations. Curr Treat Options Oncol. 2020;21(9):72.
Zhang M, Ma W, Liu H, Jiang Y, Qin L, Li W, et al. Osimertinib improves overall survival in patients with leptomeningeal metastases associated with egfr-mutated non-small-cell lung cancer regardless of cerebrospinal fluid T790M mutational status. Evid Based Complement Altern Med eCAM. 2021;2021:6968194.
Niu H, Zhou J, Maan H, Markman M, Niu J. Treatment of leptomeningeal metastases in a patient with non-small cell lung cancer harboring EGFR T790M mutation. Case Rep Oncol. 2017;10(3):840–5.
Hendriks LEL, Bootsma G, Mourlanette J, Henon C, Mezquita L, Ferrara R, et al. Survival of patients with non-small cell lung cancer having leptomeningeal metastases treated with immune checkpoint inhibitors. Eur J Cancer. 2019;116:182–9.
How J, Mann J, Laczniak AN, Baggstrom MQ. Pulsatile erlotinib in EGFR-positive non-small-cell lung cancer patients with leptomeningeal and brain metastases: review of the literature. Clin Lung Cancer. 2017;18(4):354–63.
Wang Y, Liu S, Wei X, Yan B, Li J, Su Z, et al. Non-small cell lung cancer leptomeningeal metastases treated with intrathecal therapy plus osimertinib and temozolomide and whole-brain radiation therapy: a case report. Onco Targets Ther. 2018;11:4733–8.
Carmona-Bayonas A, Gomez D, Martinez de Castro E, Perez Segura P, Munoz Langa J, Jimenez-Fonseca P, et al. A snapshot of cancer-associated thromboembolic disease in 2018–2019: first data from the TESEO prospective registry. Eur J Internal Med. 2020;78:41–9.
Shimizu Y, Okada K, Adachi J, Abe Y, Narumi R, Uchibori K, et al. GSK3 inhibition circumvents and overcomes acquired lorlatinib resistance in ALK-rearranged non-small-cell lung cancer. NPJ Precision Oncol. 2022;6(1):16.
Xin XL, Wang GD, Han R, Jiang Y, Liu C, Liu LS, et al. Mechanism underlying the effect of Liujunzi decoction on advanced-stage non-small cell lung cancer in patients after first-line chemotherapy. J Tradit Chin Med Chung i tsa chih ying wen pan. 2022;42(1):108–15.
Vismara M, Reduzzi C, Silvestri M, Murianni F, Lo Russo G, Fortunato O, et al. Single-cell phenotypic and molecular characterization of circulating tumor cells isolated from cryopreserved peripheral blood mononuclear cells of patients with lung cancer and sarcoma. Clin Chem. 2022;68:691–701.
Natarajan SR, Ponnusamy L, Manoharan R. MARK2/4 promotes Warburg effect and cell growth in non-small cell lung carcinoma through the AMPKalpha1/mTOR/HIF-1alpha signaling pathway. Biochim Biophys Acta. 2022;1869(7): 119242.
Zheng Y, Tang L, Liu Z. Multi-omics analysis of an immune-based prognostic predictor in non-small cell lung cancer. BMC Cancer. 2021;21(1):1322.
Zhu W, Shi L, Gong Y, Zhuo L, Wang S, Chen S, et al. Upregulation of ADAMDEC1 correlates with tumor progression and predicts poor prognosis in non-small cell lung cancer (NSCLC) via the PI3K/AKT pathway. Thorac Cancer. 2022;13:1027–39.
Dong Y, Tian J, Yan B, Lv K, Li J, Fu D. Liver-metastasis-related genes are potential biomarkers for predicting the clinical outcomes of patients with pancreatic adenocarcinoma. Pathol Oncol Res POR. 2021;27:1609822.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Saboundji K, Auliac JB, Perol M, Francois G, Janicot H, Marcq M, et al. Efficacy of osimertinib in EGFR-mutated non-small cell lung cancer with leptomeningeal metastases pretreated with EGFR-tyrosine kinase inhibitors. Target Oncol. 2018;13(4):501–7.
Li K, Guo S, Tong S, Sun Q, Jin S, Qi B, et al. Identification of keratinocyte differentiation-involved genes for metastatic melanoma by gene expression profiles. Comput Math Methods Med. 2021;2021:9652768.
Sigin VO, Kalinkin AI, Kuznetsova EB, Simonova OA, Chesnokova GG, Litviakov NV, et al. DNA methylation markers panel can improve prediction of response to neoadjuvant chemotherapy in luminal B breast cancer. Sci Rep. 2020;10(1):9239.
Qiao J, Cui SJ, Xu LL, Chen SJ, Yao J, Jiang YH, et al. Filamin C, a dysregulated protein in cancer revealed by label-free quantitative proteomic analyses of human gastric cancer cells. Oncotarget. 2015;6(2):1171–89.
Wu M, Michaud EJ, Johnson DK. Cloning, functional study and comparative mapping of Luzp2 to mouse chromosome 7 and human chromosome 11p13-11p14. Mamm Genome Off J Int Mamm Genome Soc. 2003;14(5):323–34.
Stepanov V, Vagaitseva K, Bocharova A, Marusin A, Markova V, Minaycheva L, et al. Analysis of association of genetic markers in the LUZP2 and FBXO40 genes with the normal variability in cognitive performance in the elderly. Int J Alzheimer’s Disease. 2018;2018:2686045.
Zhao J, Zhao Y, Wang L, Zhang J, Karnes RJ, Kohli M, et al. Alterations of androgen receptor-regulated enhancer RNAs (eRNAs) contribute to enzalutamide resistance in castration-resistant prostate cancer. Oncotarget. 2016;7(25):38551–65.
Wang F, Hu YL, Feng Y, Guo YB, Liu YF, Mao QS, et al. High-level expression of PRSS3 correlates with metastasis and poor prognosis in patients with gastric cancer. J Surg Oncol. 2019;119(8):1108–21.
Raczkowski HL, Xu LS, Wang WC, DeKoter RP. The E26 Transformation-Specific-family transcription factor Spi-C is dynamically regulated by external signals in B cells. bioRxiv. 2021:2021.08.25.457658.
Khanna P, Lee JS, Sereemaspun A, Lee H, Baeg GH. GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signaling. Sci Rep. 2018;8(1):9511.
Khanna P, Chua PJ, Wong BSE, Yin C, Thike AA, Wan WK, et al. GRAM domain-containing protein 1B (GRAMD1B), a novel component of the JAK/STAT signaling pathway, functions in gastric carcinogenesis. Oncotarget. 2017;8(70):115370–83.
Rana M, Kansal R, Chaib M, Teng B, Morrrison M, Hayes DN, et al. The pancreatic cancer immune tumor microenvironment is negatively remodeled by gemcitabine while TGF-beta receptor plus dual checkpoint inhibition maintains antitumor immune cells. Mol Carcinog. 2022;61:549–57.
Wang Y, Chen R, Wa Y, Ding S, Yang Y, Liao J, et al. Tumor immune microenvironment and immunotherapy in brain metastasis from non-small cell lung cancer. Front Immunol. 2022;13: 829451.
Jiang WY, Lei QY, Liu SS, Yang L, Yang B, Zhang Y. The effect of HOXC10 gene on biological behaviors of glioma cells and mechanism in tumor microenvironment. Zhonghua zhong liu za zhi [Chinese journal of oncology]. 2022;44(3):228–37.
Kim CG, Kim G, Kim KH, Park S, Shin S, Yeo D, et al. Distinct exhaustion features of T lymphocytes shape the tumor-immune microenvironment with therapeutic implication in patients with non-small-cell lung cancer. J Immunother Cancer. 2021;9(12):e002780.
Guo Q, Xiao XY, Wu CY, Li D, Chen JL, Ding XC, et al. Clinical roles of risk model based on differentially expressed genes in mesenchymal stem cells in prognosis and immunity of non-small cell lung cancer. Front Genet. 2022;13: 823075.
Lee H, Choi H. Investigating the clinico-molecular and immunological evolution of lung adenocarcinoma using pseudotime analysis. Front Oncol. 2022;12: 828505.
Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol. 2018;13:395–412.
Siersbaek R, Scabia V, Nagarajan S, Chernukhin I, Papachristou EK, Broome R, et al. IL6/STAT3 signaling hijacks estrogen receptor alpha enhancers to drive breast cancer metastasis. Cancer Cell. 2020;38(3):412–23 e9.
Acknowledgements
We sincerely acknowledge the contributions from the UCSC Xena website. UCSC Xena (xenabrowser.net).
Funding
This study was supported by the Project Agreement for Science & Technology Development, Jilin Province (Project No. 20200404060YY).
Author information
Authors and Affiliations
Contributions
All authors participated in the design, interpretation of the studies and analysis of the data and review of the manuscript; HD, LS and ZC have equal contributions, HD, ZC and LS wrote this paper. HX and XD provided experimental concepts and design, YZ offered scientific direction, and reviewed the manuscript. PC and YL participated in data analysis and drew the figures. LS contributed to the conception, project administration, funding acquisition of the study and supervision, writing—review & editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ding, H., Shi, L., Chen, Z. et al. Construction and evaluation of a prognostic risk model of tumor metastasis-related genes in patients with non-small cell lung cancer. BMC Med Genomics 15, 187 (2022). https://doi.org/10.1186/s12920-022-01341-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01341-6