
Abstract
Background
Tyrosine hydroxylase deficiency (THD) is a rare movement disorder with broad phenotypic expression caused by bi-allelic mutations in the TH gene, which encode for tyrosine hydroxylase (TH) protein. Some patients with THD have improvement in dystonia with carbidopa–levodopa, a synthetic form of dopamine typically used in Parkinson’s disease, and are considered to have dopa-responsive THD. THD has been found in 0.5–1 per million persons, although due to overlapping symptoms with other disorders and the need for genetic testing, prevalence is likely underestimated. Existing literature describes some patients with THD having intellectual disability, but comorbid autism spectrum disorder (ASD) has not been reported.
Case presentation
A nearly 3-year-old boy was referred to pediatric neurology due to hypotonia, delayed motor milestones, and expressive speech delay. Whole exome sequencing confirmed tyrosine hydroxylase deficiency, detecting a novel variant p.S307C first reported here. The child was treated with carbidopa–levodopa with an excellent response, resulting in improved balance, fewer falls, and improved ability to jump, run and climb stairs. He was determined to have dopa-responsive THD. Due to his delays in expressive speech, the boy also had an assessment with a developmental and behavioral pediatrician, who identified a pattern of social pragmatic speech delay, sensory sensitivities, and restricted interests, and determined that he met criteria for a diagnosis of ASD.
Conclusions
While ASD can stand alone as a clinical diagnosis, it is also a cardinal feature of other genetically-based neurological disorders. To our knowledge, this is the first case that describes a patient with both disorders. Perhaps THD may be among the genetic disorders linked with ASD.
Similar content being viewed by others


Background
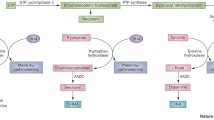
Tyrosine Hydroxylase Deficiency (THD) is a rare genetic disorder caused by bi-allelic mutations in the TH gene, which encode for tyrosine hydroxylase (TH) protein [1, 2]. Tyrosine hydroxylase catalyzes the conversion of l-tyrosine to l-dihydroxyphenylalanine (l-DOPA or levodopa), which is a rate-limiting step in the biosynthesis of dopamine, norepinephrine and epinephrine [3, 4]. Dopaminergic neurons are located in the substantia nigra and ventral tegmental areas of the midbrain, and projects to the basal ganglia for motor coordination, limbic system for emotion and memory formation, and the prefrontal cortex where it is involved in motor planning and cognition [5, 6]. Dopamine deficiency of any etiology can impair these processes [5,6,7]. THD is also known as autosomal recessive dopa-responsive dystonia. The dopa-responsive dystonias are a group of genetic syndromes caused by defects in the dopamine synthesis pathway, often associated with a therapeutic response to levodopa therapy, although some patients have lack of response or adverse responses to levodopa therapy. Three phenotypically distinct syndromes have been described based on symptoms and response to levodopa therapy. From mild to severe, they are: (1) TH-deficient dopa-responsive dystonia, (2) TH-deficient infantile parkinsonism with motor delay, and (3) TH-deficient progressive infantile encephalopathy. Mild cases may present later in childhood with lower limb dystonia and difficulty walking, worsening towards the end of the day, i.e. diurnal fluctuation [8, 9]. Dopa-responsive dystonias are estimated to have a population prevalence of 0.5–1 per million worldwide [8, 10]. Similar to other dopa-responsive dystonias, persons with THD have improvement in dystonia with carbidopa–levodopa, a synthetic form of dopamine typically used in Parkinson’s disease [8, 10]. Severe cases may present as infantile parkinsonism (hypokinesia, rigidity of extremities, tremor), with markedly delayed motor milestones and often truncal hypotonia. The most severe form, progressive infantile encephalopathy, has a much earlier onset (before 3–6 months), and may present with fetal distress, severe hypokinesia, limb hypertonia, hyperreflexia, truncal hypotonia, paroxysmal periods of lethargy alternating with irritability [11], and increased likelihood of behavioral problems and/or intellectual disabilities [8, 9]. Currently, we estimate there are over 100 reports of THD in the medical literature [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. However, due to THD’s phenotypic heterogeneity and overlap with other disorders, genetic testing is necessary to confirm the diagnosis, and thus accurate estimates of population prevalence are difficult to obtain [8, 11].
In addition to these neurologic manifestations, intellectual disability has been described in cases of THD, most commonly in patients with very severe forms of THD [8, 11]. To our knowledge, there are currently no reports of autism spectrum disorder (ASD) in children with THD.
In this case report, we describe a young child presenting with delays in development, ultimately diagnosed with both THD and ASD.
Case presentation
A nearly 3-year-old boy was referred to pediatric neurology after his Early Intervention physical therapist and his primary pediatrician found significant delays in gross motor, adaptive and social skills. Due to COVID-19, his therapies were interrupted for almost a year, and when re-initiated, his parents were most concerned about his balance; he would fall 10–20 times per day which is atypical. Overall, his development was delayed across domains: he rolled at 7 months and sat without support at 8 months. For reference, most children roll by 6 months of age and sit without support by 9 months of age [39]. Although he had first words just before a year of age, at 33 months his expressive language skills were delayed with mostly only 2-word sentences. Most children by 3 years of age are able to engage in back-and-forth conversation and answer simple questions [39]. He was also reported to have texture sensitivities, for example refusing all mushy foods. The child was born via spontaneous vaginal birth at term after an unremarkable pregnancy with Apgars of 6 and 8. He had a brief NICU course due to respiratory distress syndrome and was found to have a muscular VSD and bicuspid aortic valve. On the neurologist’s physical exam, the patient’s height, weight and head circumference were above the 99th percentile for age, per CDC standards. There were no dysmorphic features. The cranial nerve exam, including extraocular eye movements, was normal, with the exception of drooling. He had marked diffuse axial and appendicular hypotonia, without spasticity, and with normal muscle bulk. Strength was difficult to assess, however, the patient rose from the floor without assistance or Gower’s maneuver, and opened a heavy exam room door. The deep tendon reflexes were normal, with the exception of two beats of symmetrical ankle clonus. The patient energetically explored the exam room, and there was no dysmetria when reaching for objects. The gait was wide-based, with arms held in high-guard versus dystonic posture. The patient could run, but fell frequently without using his arms to brace for the fall. Family history was unremarkable for similarly affected relatives. Mother reported a history of an Individual Education Plan in school for reading and as an adult idiopathic intracranial hypertension that was treated with medication. Father reported attention deficit hyperactivity disorder but was otherwise healthy. Patient’s younger sibling was reported healthy, with normal development. There was no reported family history of neurodevelopmental disability. The maternal ancestry was Irish and German and paternal ancestry was Polish and German. Parents were non-consanguineous.
The initial diagnostic evaluation included blood tests (creatine kinase, lactate, thyroid stimulating hormone, plasma amino acids and acylcarnitine profile) and an MRI brain which were all normal (Table 1). The ongoing differential was broad, including muscle dystrophies, lysosomal storage disease, and congenital myasthenic syndromes. Given complex picture and broad differential whole exome sequencing (WES) was recommended. Meanwhile, the child’s pediatric neurologist referred him to a developmental and behavioral pediatrician to evaluate his expressive speech delay. The WES was performed at GeneDx, using buccal samples from patient and both parents. Massively parallel (NextGen) sequencing was done on an Illumina system and reads were aligned to human genome build GRCh37/UCSC hg19. The mean depth of coverage was 121×. Sequence variants are classified based on ACMG/AMP guidelines [40]. The WES revealed the patient to be compound heterozygous for two variants in TH gene: c.698G > A; p.R233H & c.920 C > G; p.S307C (NM_199292.2 confirming a diagnosis of autosomal recessive THD. R233H transversion is the most frequently seen pathogenic variant in individuals with THD, carried by 42 patients reported thus far in the literature [41]. The S307C variant has not previously been reported to our knowledge and is a novel variant. Authors of this publication did not conduct functional studies of the p.S307C variant. In silico analysis supports that this missense variant has a deleterious effect on protein structure/function. Provean score for this variant was − 4.83. The variant was classified as likely pathogenic. Parental testing confirmed these variants were inherited; sibling tested negative for both TH variants. No variants of unknown significance or variants in other candidate genes were identified on WES.
The patient’s phenotype was considered to be mild to moderate given his age of onset, ataxia, gait, developmental delays, and lack of parkinsonian features. The patient was titrated to a final dose of carbidopa–levodopa (SINEMET) 25–100 mg tablet; ¼ tablet TID. No side effects were reported. The patient had an excellent response, resulting in improved balance, fewer falls, and improved ability to jump, run and climb stairs. These improvements confirmed that the patient had the dopa-responsive form of THD.
At 3.5 years of age, the patient presented for assessment by a developmental and behavioral pediatrician. At this time the patient had a confirmed THD diagnosis, but was not yet being treated with Carbidopa–levodopa therapy. The Developmental and Behavioral Pediatrician noted a presentation concerning for ASD: intermittent eye contact which was not sustained, difficulty with conversational reciprocity, restricted interest in letters and numbers, and sensory issues with feeding. He also had a developmental history consistent with ASD, delay in expressive language with poor social pragmatic skills (at 2 years of age he had only about 20 words which he used for labeling versus requesting or sharing). He was assessed formally with an Autism Diagnostic Observation Schedule-Module 2, which confirmed the diagnosis of ASD. The patient was referred to Applied Behavioral Analytic Therapy, a standard of care intervention for children with ASD where clinicians utilize behavior principles to (1) promote socially significant behavioral changes, (2) improve the patient’s overall adaptive behavior skills, (3) develop language and cognitive skills, and (4) decrease maladaptive behavior associated with ASD’s core symptoms. The patient was also recommended to have a chromosomal microarray and Fragile X testing, which were not performed.
Discussion and conclusion
In this case report, we present a young boy with gross motor delays, characterized by hypotonia, dystonia and ataxia, and delays in social pragmatic speech. He was diagnosed with levodopa-responsive THD and ASD. To our knowledge, existing reports of THD have described comorbid intellectual disability but not ASD, however our case raises the possibility that THD causes or increases risk for ASD.
ASD is a complex neurodevelopmental diagnosis with clear genetic risk, based on studies of recurrence rates in siblings and twins [42,43,44,45], without a singular genetic etiology. While ASD can stand alone as a clinical diagnosis, it is also a cardinal feature of other genetically-based neurological disorders such as Fragile X Disorder (30–54% of males affected) [46,47,48,49,50,51,52], Tuberous Sclerosis Complex (25–50% of individuals affected) [53, 54] and Angelman Syndrome (~ 30% of individuals affected) [55]. Although ASD is a common neurodevelopmental disorder increasing in prevalence, now diagnosed in 1 in 44 children [56], it may also be possible that genetic variants in the TH gene may be associated with ASD. Unfortunately one of the limitations of this case is the yet unavailable information on the patient’s chromosomal microarray and Fragile X testing results, which are currently other recommended genetic tests when a child is diagnosed with ASD [57].
The prevalence of ASD has been steadily increasing over the past few decades [58,59,60,61]. One proposed cause for this increase is the greater recognition of ASD by health providers and families. Another is the growing recognition that ASD presents in a wide range of symptoms in individuals with varying levels of ability, coupled with improvements to formal diagnostic criteria systems (ICD-10; DSM-IV) [62]. This second point has led to a greater appreciation for the distinction between ASD and intellectual disability, though they are often co-occurring. Diagnostic substitution plays a role in explaining increasing ASD prevalence (and decreasing intellectual disability prevalence) [63, 64], however other factors are at play.
This patient, presenting with hypotonia, frequent falls, delays in social communication, and sensory atypicalities was found to have THD and ASD diagnoses. To our knowledge, this is the first case that describes a patient with both disorders and a novel TH variant (S307C). Perhaps THD may be among the genetic disorders linked with ASD and potential ASD diagnoses ought to be considered when diagnosing a child with THD who also has challenges with social communication.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- THD:
-
Tyrosine hydroxylase deficiency
- TH:
-
Tyrosine hydroxylase
- l-DOPA:
-
l-Dihydroxyphenylalanine
- ASD:
-
Autism spectrum disorder
- WES:
-
Whole exome sequencing
References
Kliegman R, Geme JS. Nelson textbook of pediatrics. 21st ed. Philadelphia: Elsevier; 2019.
Dong HY, Feng JY, Yue XJ, Shan L, Jia FY. Dopa-responsive dystonia caused by tyrosine hydroxylase deficiency: three cases report and literature review. Medicine (Baltimore). 2020;99(33):e21753.
Nagatsu T, Levitt M, Udenfriend S. Tyrosine hydroxylase. The initial step in norepinephrine biosynthesis. J Biol Chem. 1964;239:2910–7.
Szigetvari PD, Muruganandam G, Kallio JP, Hallin EI, Fossbakk A, Loris R, et al. The quaternary structure of human tyrosine hydroxylase: effects of dystonia-associated missense variants on oligomeric state and enzyme activity. J Neurochem. 2019;148(2):291–306.
Klein MO, Battagello DS, Cardoso AR, Hauser DN, Bittencourt JC, Correa RG. Dopamine: functions, signaling, and association with neurological diseases. Cell Mol Neurobiol. 2019;39(1):31–59.
Speranza L, di Porzio U, Viggiano D, de Donato A, Volpicelli F. Dopamine: the neuromodulator of long-term synaptic plasticity, reward and movement control. Cells. 2021;10(4):735.
Nygaard G, Szigetvari PD, Grindheim AK, Ruoff P, Martinez A, Haavik J, et al. Personalized medicine to improve treatment of dopa-responsive dystonia—a focus on tyrosine hydroxylase deficiency. J Pers Med. 2021;11(11):1186.
Furukawa Y, Kish S, et al. Tyrosine hydroxylase deficiency. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews((R)). Seattle: University of Washington; 2008. (Updated 2017 May 11).
Katus LE, Frucht SJ. An unusual presentation of tyrosine hydroxylase deficiency. J Clin Mov Disord. 2017;4:18.
Swoboda KJ, Walker MA, et al. Neurotransmitter-related disorders. In: Swaiman KF, Ashwal S, Ferriero DM, Schor NF, Finkel RS, Gropman AL, et al., editors. Swaiman’s pediatric neurology. 6th ed. Amsterdam: Elsevier; 2017. p. 355–61.
Willemsen MA, Verbeek MM, Kamsteeg EJ, de Rijk-van Andel JF, Aeby A, Blau N, et al. Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis. Brain. 2010;133(Pt 6):1810–22.
Hubschmann OK, Horvath G, Cortes-Saladelafont E, Yildiz Y, Mastrangelo M, Pons R, et al. Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines. Nat Commun. 2021;12(1):5529.
de Rijk-van Andel JF, Gabreels FJM, Geurtz B, Steenbergen-Spanjers GCH, van den Heuvel LPWJ, Smeitink JAM, et al. L-dopa-responsive infantile hypokinetic rigid parkinsonism due to tyrosine hydroxylase deficiency. Neurology. 2000;55(12):1926–8.
Schiller A, Wevers RA, Steenbergen GC, Blau N, Jung HH. Long-term course of L-dopa-responsive dystonia caused by tyrosine hydroxylase deficiency. Neurology. 2004;63(8):1524–6.
Chi CS, Lee HF, Tsai CR. Tyrosine hydroxylase deficiency in Taiwanese infants. Pediatr Neurol. 2012;46(2):77–82.
Hou M, Yang C, Hu J, Guo Y, Liu P, Liu Y, et al. Levodopa is effective in the treatment of three Chinese tyrosine hydroxylase (TH) deficiency children. Int J Dev Neurosci. 2019;78:28–32.
Goswami JN, Sankhyan N, Singhi PD. An Indian family with tyrosine hydroxylase deficiency. Indian Pediatr. 2017;54(6):499–501.
Hoffmann GF, Assmann B, Brautigam C, Dionisi-Vici C, Haussler M, de Klerk JB, et al. Tyrosine hydroxylase deficiency causes progressive encephalopathy and dopa-nonresponsive dystonia. Ann Neurol. 2003;54(Suppl 6):S56-65.
Wang Y, Wang C, Liu M, Xu W, Wang S, Yuan F, et al. Segawa syndrome caused by TH gene mutation and its mechanism. Front Genet. 2022;13:1004307.
Pons R, Serrano M, Ormazabal A, Toma C, Garcia-Cazorla A, Area E, et al. Tyrosine hydroxylase deficiency in three Greek patients with a common ancestral mutation. Mov Disord. 2010;25(8):1086–90.
Zafeiriou DI, Willemsen MA, Verbeek MM, Vargiami E, Ververi A, Wevers R. Tyrosine hydroxylase deficiency with severe clinical course. Mol Genet Metab. 2009;97(1):18–20.
Szentivanyi K, Hansikova H, Krijt J, Vinsova K, Tesarova M, Rozsypalova E, et al. Novel mutations in the tyrosine hydroxylase gene in the first Czech patient with tyrosine hydroxylase deficiency. Prague Med Rep. 2012;113(2):136–46.
Feng B, Sun G, Kong Q, Li Q. Compound heterozygous mutations in the TH gene in a Chinese family with autosomal-recessive dopa-responsive dystonia: a case report. Medicine (Baltimore). 2018;97(44):e12870.
Janssen E, Oosterloo M, Rubio-Gozalbo E, van Gassen K, Nicolai J. Teaching video neuroimage: improvement in motor development after start of levodopa in tyrosine hydroxylase deficiency. Neurology. 2021;97(5):e540.
Leuzzi V, Mastrangelo M, Giannini MT, Carbonetti R, Hoffmann GF. Neuromotor and cognitive outcomes of early treatment in tyrosine hydroxylase deficiency type B. Neurology. 2017;88(5):501–2.
Brautigam C, Steenbergen-Spanjers GC, Hoffmann GF, Dionisi-Vici C, van den Heuvel LP, Smeitink JA, et al. Biochemical and molecular genetic characteristics of the severe form of tyrosine hydroxylase deficiency. Clin Chem. 1999;45(12):2073–8.
Moller LB, Romstad A, Paulsen M, Hougaard P, Ormazabal A, Pineda M, et al. Pre- and postnatal diagnosis of tyrosine hydroxylase deficiency. Prenat Diagn. 2005;25(8):671–5.
Panda S, Jain S, Dholakia D, Uppilli BR, Faruq M. Prolonged episodic dystonia in tyrosine hydroxylase deficiency due to homozygous c.698G>A (p.Arg233His) mutation-A diagnostic challenge. Mov Disord Clin Pract. 2022;9(8):1136–9.
Champagne M, Horvath GA, Perreault S, Gauthier J, Hyland K, Soucy JF, et al. Intermittent neurologic decompensation: an underrecognized presentation of tyrosine hydroxylase deficiency. JIMD Rep. 2022;63(5):400–6.
Hull M, Emrick L, Sadat R, Parnes M. A case of treatable encephalopathy, developmental regression, and proximal tremor. Parkinsonism Relat Disord. 2021;93:111–3.
Giovanniello T, Claps D, Carducci C, Carducci C, Blau N, Vigevano F, et al. A new tyrosine hydroxylase genotype associated with early-onset severe encephalopathy. J Child Neurol. 2012;27(4):523–5.
Giovanniello T, Leuzzi V, Carducci C, Carducci C, Sabato ML, Artiola C, et al. Tyrosine hydroxylase deficiency presenting with a biphasic clinical course. Neuropediatrics. 2007;38(4):213–5.
Bijarnia-Mahay S, Jain V, Thony B. Tyrosine hydroxylase deficiency-clinical insights and a novel deletion in TH gene in an Indian patient. JIMD Rep. 2020;53(1):12–5.
De Lonlay P, Nassogne MC, van Gennip AH, van Cruchten AC, BillattedeVillemeur T, Cretz M, et al. Tyrosine hydroxylase deficiency unresponsive to L-dopa treatment with unusual clinical and biochemical presentation. J Inherit Metab Dis. 2000;23(8):819–25.
Grattan-Smith PJ, Wevers RA, Steenbergen-Spanjers GC, Fung VS, Earl J, Wilcken B. Tyrosine hydroxylase deficiency: clinical manifestations of catecholamine insufficiency in infancy. Mov Disord. 2002;17(2):354–9.
Cassani E, Barichella M, Ferri V, Pusani C, Goldwurm S, Siri C, et al. Protein-redistribution diet in a case of tyrosine hydroxylase enzyme deficiency. Mov Disord. 2017;32(5):794–5.
Haussler M, Hoffmann GF, Wevers RA. L-dopa and selegiline for tyrosine hydroxylase deficiency. J Pediatr. 2001;138(3):451–2.
Dionisi-Vici C, Hoffmann GF, Leuzzi V, Hoffken H, Brautigam C, Rizzo C, et al. Tyrosine hydroxylase deficiency with severe clinical course: clinical and biochemical investigations and optimization of therapy. J Pediatr. 2000;136(4):560–2.
Centers for Disease Control and Prevention (CDC), National Center on Birth Defects and Developmental Disabilities (NCBDDD). Developmental milestone checklists for WIC 2022 [Available from: https://www.cdc.gov/ncbddd/actearly/pdf/FULL-LIST-CDC_LTSAE-Checklists2021_Eng_FNL2_508.pdf.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Yao CM, Deng YX, Wang YJ, Gao BQ, Zhao CS. R233H mutation in patients with tyrosine hydroxylase deficiency and corresponding phenotypes: a study of four cases and literature review. J Integr Neurosci. 2022;21(1):35.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68(11):1095–102.
Taylor MJ, Rosenqvist MA, Larsson H, Gillberg C, D’Onofrio BM, Lichtenstein P, et al. Etiology of autism spectrum disorders and autistic traits over time. JAMA Psychiat. 2020;77(9):936–43.
Tick B, Bolton P, Happe F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry. 2016;57(5):585–95.
Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311(17):1770–7.
Yu TW, Berry-Kravis E. Autism and fragile X syndrome. Semin Neurol. 2014;34(3):258–65.
Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37(4):738–47.
Hall SS, Lightbody AA, Reiss AL. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am J Ment Retard. 2008;113(1):44–53.
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, et al. Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008;113(6):427–38.
Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, et al. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A(3):225–34.
Budimirovic DB, Bukelis I, Cox C, Gray RM, Tierney E, Kaufmann WE. Autism spectrum disorder in Fragile X syndrome: differential contribution of adaptive socialization and social withdrawal. Am J Med Genet A. 2006;140A(17):1814–26.
Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE. Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A. 2009;149A(6):1125–37.
Wiznitzer M. Autism and tuberous sclerosis. J Child Neurol. 2004;19(9):675–9.
de Vries P, Humphrey A, McCartney D, Prather P, Bolton P, Hunt A, et al. Consensus clinical guidelines for the assessment of cognitive and behavioural problems in Tuberous Sclerosis. Eur Child Adolesc Psychiatry. 2005;14(4):183–90.
Richards C, Jones C, Groves L, Moss J, Oliver C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909–16.
Maenner MJ, Shaw KA, Bakian AV, Bilder DA, Durkin MS, Esler A, et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ. 2021;70(11):1–16.
Schaefer GB, Mendelsohn NJ, Practice P. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. 2013;15(8):399–669.
Zeidan J, Fombonne E, Scorah J, Ibrahim A, Durkin MS, Saxena S, et al. Global prevalence of autism: a systematic review update. Autism Res. 2022;15(5):778–90.
Newschaffer CJ, Falb MD, Gurney JG. National autism prevalence trends from United States special education data. Pediatrics. 2005;115(3):e277–82.
Fombonne E. The prevalence of autism. JAMA. 2003;289(1):87–9.
Autism, Developmental Disabilities Monitoring Network Surveillance Year Principal I, Centers for Disease C. Prevalence of autism spectrum disorders–autism and developmental disabilities monitoring network, 14 sites, United states, 2008. MMWR Surveill Summ. 2012;61(3):1–19.
Hansen SN, Schendel DE, Parner ET. Explaining the increase in the prevalence of autism spectrum disorders: the proportion attributable to changes in reporting practices. JAMA Pediatr. 2015;169(1):56–62.
Howlin P. Autism and diagnostic substitution. Dev Med Child Neurol. 2008;50(5):325.
Shattuck PT. The contribution of diagnostic substitution to the growing administrative prevalence of autism in US special education. Pediatrics. 2006;117(4):1028–37.
Acknowledgements
We would like to acknowledge this patient and his family, who gave the corresponding author (SS) permission for his rare case to be presented to the medical literature, and provided consent for care providers at different institutions to work together and share his records.
Funding
Dr. Sobotka receives support from The Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, K23 HD097276) and the T73 Leadership Education in Neurodevelopmental and Related Disorders Training Program (LEND, T73MC11047). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SS was the Developmental and Behavioral Pediatrician treating this patient, she conceptualized the case report, and made substantial contributions to drafting and editing the manuscript. ZR completed the initial literature review and was a contributor to the drafting and editing of the manuscript. EL contributed to the literature review, drafting and editing the manuscript. JH was the Pediatric Neurologist and contributed to drafting and editing the manuscript. LDS was the Genetic Counselor and contributed to drafting and editing the manuscript. All authors read and approved the final manuscript. All authors have agreed to be personally accountable for their own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and the family provided release of information to collate medical record data for multiple health systems.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Reyes, Z.M.D., Lynch, E., Henry, J. et al. Diagnosis of autism in a rare case of tyrosine hydroxylase deficiency: a case report. BMC Med Genomics 16, 78 (2023). https://doi.org/10.1186/s12920-023-01510-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01510-1