
Abstract
Background
Rhizomelic limb shortening with dysmorphic features (RLSDF) has already been a disorder of the rare autosomal recessive skeletal dysplasia, just having a few reported cases. RLSDF is caused by protein kinase domain containing, cytoplasmic(PKDCC)gene variants. In this study, we describe the clinical features and potential RLSDF molecular etiology in a fetus from China.
Methods
Genomic DNA (gDNA) extracted from the fetal muscle tissue and parents’ peripheral blood was subjected to chromosomal microarray analysis (CMA) and trio-based whole exome sequencing (Trio-WES). The candidate pathogenic variants were verified by using Sanger sequencing.
Results
Trio-WES identified two compound heterozygous variants in PKDCC, c.346delC (p.Pro117Argfs*113) and c.994G > T (p.Glu332Ter), inherited from the father and mother, respectively. Both variants are classified as pathogenic according to American College of Medical Genetics and Genomics guidelines.
Conclusions
We reported the first prenatal case of RLSDF caused by PKDCC in the Chinese population. Our findings extended the variation spectrum of PKDCC and emphasized the necessity of WES for the early diagnosis of skeletal dysplasia and other ultrasound structural abnormalities in fetuses.
Similar content being viewed by others


Background
Rhizomelic limb shortening with dysmorphic features (RLSDF, OMIM#618,821) is a rare autosomal recessive disorder characterized of rhizomelic shortening of the lower and upper limbs and changeable dysmorphic features, containing short neck, prominent forehead, macrocephaly, broad or depressed nasal bridge, micrognathia, along with long philtrum. Other features included obesity, mild plagiocephaly, laryngomalacia, short thumbs, mild bilateral conductive hearing loss, acanthosis nigricans, central hypotonia and mildly delayed myelination [1, 2]. This disease results from biallelic variants in the protein kinase domain containing, cytoplasmic (PKDCC) gene, which is located in chromosome 2p21 and contains seven exons. It encodes an integrated component of Hedgehog signaling demanded for normal chondrogenesis and bone development [2]. Studies on mice suggest that Hedgehog signaling can be regulated by PKDCC (also known as Vertebrate lonesome kinase, VLK) [3]. The Hedgehog signalling regulation is essential in the course of the bone development and repair [4]. PKDCC null mice show shorted long bones due to delayed endochondral ossification, as well as craniofacial abnormalities, including shortened and small nasal capsule and maxilla [5, 6].
To date, only nine families with molecularly confirmed variants of the PKDCC gene have been reported [1, 2]. The genotype-phenotype correlation of RLSDF is still unclear [2]. There is no specific treatment for RLSDF. The life expectancy of individuals with RLSDF is usually unaffected. In this study, we reported a female fetus with skeletal dysplasia through RLSDF, wherein we identified compound heterozygous variants affecting the PKDCC gene. To the best of our knowledge, this is the earliest identification of RLSDF by morphology ultrasound and the first report of PKDCC variants in a prenatal case of RLSDF.
Materials and methods
Clinical report
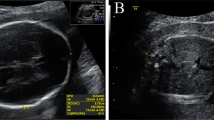
The parents of the female fetus were nonconsanguineous and healthy. This was their first pregnancy (Fig. 1A). The pregnant woman conceived and was not exposed to poison or radioactive substances. The 22+ 1-week morphology ultrasound demonstrated rhizomelia of the upper limbs; the echoes of the spine and other bones slightly weakened (Fig. 1B-E). The length of the humerus and ulna was 27 mm (Table 1). An assumed diagnosis of skeletal dysplasia was made. Nevertheless, an accurate diagnosis was challenging to fulfill based upon the limited ultrasound discoveries. No other abnormalities were observed. The pregnant woman and her husband both showed no family history of skeletal dysplasia disorders or congenital malformations. Based on the ultrasound findings, the pregnancy was terminated 3 weeks later. The female fetus weighed 750 g with a height of 23 cm. Dysmorphic features were apparent with rhizomelia of the upper limbs, prominent forehead, and nasal planus (Fig. 1F). As for the exterior examination, its remainder was normal. Chromosomal microarray analysis (CMA) and trio-based whole exome sequencing (WES) analysis were conducted using the muscular tissue of the fetus and parents’ blood.

A Pedigree of the family with RLSDF. B-E Morphology ultrasound at a gestation of 22+ 1 weeks showing short humeri and ulna; the echoes of the spine and other bones were slightly weakened. F Appearance of the fetus after termination of pregnancy at a gestation period of 25+ 3 weeks, showing proband with rhizomelia of the upper limbs, flat face, prominent forehead, and nasal planus
All participators offered the written informed consent. The protocols of this study were approved by the ethics committee of the Ningbo Women and Children’s Hospital (Zhejiang, China).
Sample collection
Genomic DNA was extracted from the muscular tissue of the fetus and peripheral blood originating in parents using the QIAamp DNA Blood Mini Kit (QIAGEN, Germany) in light of the producer’s instructions (www.qiagen. com).
Chromosomal microarray analysis
We conducted CMA using CytoScan 750 K array (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s protocol. Data was analyzed by the Chromosome Analysis Suite 4.0 (Afymetrix, Santa Clara, CA, USA). Copy number variants (CNVs) were further assessed according to the Online Mendelian Inheritance in Man (OMIM), ClinVar, Database of Genomic Variants, Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER), PubMed, and other databases. The pathogenicity interpretation of CNVs was conducted according to the standards and guidelines of the American College of Medical Genetics (ACMG) and the Clinical Genome Resource (ClinGen) [7].
Whole exome sequencing (WES)
The WES of the genomic DNA of the proband and the parents was conducted with SureSelect Human All Exon V6 kits (Agilent) as well as a NovaSeq 6000 sequencer (Illumina, San Diego, CA). All indels and single nucleotide variants (SNVs) were filtrated and estimated through multiple databases, including the 1000 Genomes Project dataset, HapMap, Single-Nucleotide Polymorphism Database (dbSNP). Exome Variant Server databases were employed for the purpose of determining the pathogenicity and harmfulness of identified variations. All variants were subjected to biological effect analysis, that included the use of programs such as SIFT, PROVEAN, PolyPhen-2, Mutation Taster, deleterious annotation of genetic variants using neural networks (DANN), and Human Splicing Finder to forecast if an amino acid indel or substitution exerts a pivotal biological effect. Pathogenic variants are categorized in light of the ACMG and the Association for Molecular Pathology (AMP) [8].
Sanger sequencing verification of thePKDCCgene.
Sanger sequencing was conducted to confirm the potential causative variants from this family. Primers were devised by means of Primer 5.0. The primers are as follows: PKDCC-c.346delC, forward, 5′-TCCTCAACGTGCTCTTCGCTC-3′, reverse, 5′-GGGTTCTCTTCCAGCCAGGT-3′ and PKDCC-c.G994T, forward, 5′-AGAAGAGAAGTGCCAACCCC-3′, reverse, and 5′-TTCGATGGAGTTCCCGAGTC-3′. The sequence data was analyzed by means of Sequencing Analysis Software 6 (Applied Biosystems, Foster City, CA, USA).
Results
No abnormality was observed in the pathogenic CNVs in the fetus by CMA. WES revealed novel compound heterozygous variants in the PKDCC (NM_138370.3) gene c.346delC(p.Pro117Argfs*113) /c.994G > T(p.Glu332Ter). Neither variant was reported previously. The variant c.346delC(p.Pro117Argfs*113) of the fetus was a frameshift variant in exon 1 of the PKDCC gene, which could cause nonsense-mediated mRNA decay (NMD) in silico. We verified that this variant was inherited from her mother by Sanger sequencing. The other variant c.994G > T(p.Glu332Ter) of the PKDCC gene in the fetus was a nonsense variant and Sanger sequencing indicated that this variant was inherited from her father (Fig. 2). According to the classification guidelines for sequence variants from ACGM/AMP, the variants c.346delC (p.Pro117Argfs*113) and c.994G > T (p.Glu332Ter) in the PKDCC gene were classified to be pathogenic (PVS1, PM2_Supporting, PM3, PP4).

Sanger sequencing validation of the PKDCC variants identified by using WES. Variant c.346delC(p.Pro117Argfs*113) in the PKDCC gene in the heterozygous state (father and sibling). Variant c.994G > T(p.Glu332Ter) in the PKDCC gene in the heterozygous state (mother)
Prenatal diagnosis
Following the identification of the PKDCC variants, the parents selected for prenatal diagnostic testing through amniotic fluid sampling at a gestation period of 19+ 2 weeks in their second pregnancy. That fetus was found to only carry the maternal variant (Fig. 2), consistent with the normal ultrasound scan at 19-weeks. At birth, there was no evidence of RLSDF and after 1 year, the girl child shows normal development.
Discussion
Fetal skeletal dysplasia(SD) is one category of rare heterogeneous genetic disorder with 2.3 to 4.5 per 10,000 births [9]. In the 2019 revision of the Nosology and Classification of Genetic Skeletal Disorders, 461 types of hereditary skeleton diseases were identified, and are divided into 42 classes, containing 437 genes [10]. Most types of SD are the result of genetic variation, with a minority being associated with chromosomal anomalies, multisystem syndromes, or teratogenic environmental exposure [11]. However, the pathogenesis of more than half the SD remains unclear. Additionally, the clinical presentations of fetal SD are ambiguous, which results in a large challenge to diagnose accurately. Moreover, at present, an increasing number of molecular genetic approaches have already been adopted to prenatal diagnosis, and many diseases can be diagnosed by trio-WES based on next-generation sequencing technologies [12,13,14,15]. Prenatal genetic evaluation can provide parents more prospective information about the diagnosis, prognosis, and recurrence hazard for parents with the aim of making informed decisions and improve perinatal management [16].
In this study, we found that a female fetus with SD carried compound heterozygous PKDCC variants. We conducted WES on the fetus and her parents. We found that the compound heterozygous variants c.346delC(p.Pro117Argfs*113) and c.994G > T(p.Glu332Ter) of the PKDCC gene were in the sample of the fetus and they were inherited from her parents by Sanger sequencing, separately. This was the first reported case of RLSDF from China. Since the discovery of RLSDF at the year of 2018, 11 cases in total containing our patient have been reported [1, 2]. Most reported cases were linked to rhizomelia of the upper limb (100%), short stature (80%), hypertelorism (66.67%), flat face (66.67%), micrognathia (55.56%), short fifth digit (50%), hearing loss (50%), high and broad forehead (50%), macrocephaly (42.86%), sloping shoulders (33.33%), patellofemoral dislocation (33.33%), and prominent eyes (30%), rhizomelia of the upper limb was the most commonly reported (Table 2). Hypoplastic pituitary gland, growth hormone deficiency, and pectus excavatum were also observed in a small number of cases. In this study, the fetus showed dysmorphic characteristics and shortened long bones as the most evident morphological abnormalities. Therefore, the clinical phenotype of this fetus was basically consistent with the RLSDF. PKDCC-related SD is an extremely rare disorder. The leading skeletal characteristics of reported patients are similar, and variability was observed in the other detected phenotypes. Therefore, the prenatal evaluation of skeletal hypoplasia through ultrasound examination should lead to the consideration of genetic testing in the differential diagnosis.
The PKDCC gene was first reported by Imuta in 2009 [5]. Human PKDCC gene is 10.5 kb in size and encodes a secreted tyrosine-protein kinase with 493 amino acid residues [2]. The PKDCC is initially conveyed inside E-cadherin-positive cells however, is later limited inside E-cadherin-negative cells [6]. Currently, 11 variations in the PKDCC gene were reported to cause RLSDF in homozygous or compound heterozygous states, and the types of these variations varied, including frameshift (7/11), nonsense (2/11), missense (1/11), and splice (1/11) variants [1, 2]. This study investigated a fetus with compound heterozygous variants of PKDCC, namely, c.346delC(p.Pro117Argfs*113) and c.994G > T(p.Glu332Ter), that represent frameshift mutation and nonsense, respectively. This is consistent with the recessive inheritance pattern in this family and leads to the typical skeletal phenotype. Additionally, six of the PKDCC variants were located in exon 1, one of them in exon 2, three of them in exon 3, and one of them in intron 1 (shown in Fig. 3). We speculated that exon 1 may be a hotspot region and an important domain of the PKDCC gene. However, there is no clear correlation between genotype-phenotype of RLSDF.

Summary of all variants in the PKDCC gene (NM_138370.3) associated with RLSDF. The reported variants are indicated in red (https://www.ncbi.nlm.nih.gov/clinvar/, the last access time was April 10, 2023)
PKDCC is localized in the Golgi complex and has been associated with the longitudinal skeleton development through the regulation of chondrocyte formation [6]. During the embryonic development of mice, PKDCC is mainly expressed in condensing mesenchymal cells, with high expression in limb buds and branchial arches [5, 6, 17]. In mouse embryos, homozygous PKDCC knockout mice show deficient long bone elongation, cranial abnormalities due to the delayed formation in the shortened intestine, sternal dysgraphia, flat proliferative chondrocyte, lung hypoplasia, and cleft palate. Additionally, the newborn knockout mice died from abnormal respiration a few hours after birth [5, 6]. The Glioma-associated oncogene homolog (GLI) family zinc finger 3 (GLI3) and the Indian hedgehog (IHH) genes are members of the hedgehog pathway in bone growth. Mice lacking these genes show skeletal abnormalities, including severe dwarfism of limbs, a short body length, and craniofacial abnormalities [18,19,20,21]. Notably, PKDCC and GLI3 double knockout mice have more severe skeletal abnormalities than single knockout mice, indicating that PKDCC genetically interacts with GLI3 and also affects bone growth development through the hedgehog pathway [22]. To date, the exact pathophysiological mechanism underlying RLSDF is still not understood.
One limitation of this study is the other characteristics (except for SD and facial dysmorphism) of the fetus could not be evaluated. Although the compound heterozygous variants may impact the protein stability, this should be confirmed by functional studies. Our results emphasize the need for prenatal diagnosis. WES is useful to recognize any associated genetic disorders and provide personalized care for patients with SD and other ultrasonic structure abnormalities.
In summary, our observation of this early phenotype should help in further elucidating the function of PKDCC and its role in skeletal development. Our results have significant reference value for the molecular diagnosis of RLSDF in future studies. Our study helped expend the PKDCC pathogenic variant spectrum of RLSDF. Further functional validation is necessary to elucidate the pathogenic mechanism of the PKDCC gene in RLSDF.
Data Availability
The detected variants have been submitted to the LOVD, direct link: https://databases.lovd.nl/shared/individuals/00435461.
Abbreviations
- RLSDF:
-
Rhizomelic limb shortening with dysmorphic features
- CMA:
-
Chromosomal microarray analysis
- WES:
-
Whole exome sequencing
- CNVs:
-
Copy number variants
- SNVs:
-
Single-nucleotide polymorphism database
- SNP:
-
Single-Nucleotide Polymorphism Database
- ACMG:
-
American College of Medical Genetics and Genomics
- AMP:
-
Association for Molecular Pathology
- NMD:
-
Nonsense-mediated mRNA decay
- SD:
-
Skeletal dysplasia
- IHH:
-
Indian hedgehog
- GLI3:
-
GLI family zinc finger 3
References
Sajan SA, Ganesh J, Shinde DN, Powis Z, Scarano MI, Stone J, Winter S, Tang S. Biallelic disruption of PKDCC is associated with a skeletal disorder characterised by rhizomelic shortening of extremities and dysmorphic features. J MED GENET. 2019;56(12):850–4.
Pagnamenta AT, Belles RS, Salbert BA, Wentzensen IM, Guillen SM, Santos F, Caffo A, Ferla M, Banos-Pinero B, Pawliczak K, et al. The prevalence and phenotypic range associated with biallelic PKDCC variants. CLIN GENET. 2023;104(1):121–6.
Kim JM, Han H, Bahn M, Hur Y, Yeo CY, Kim DW. Secreted tyrosine kinase vlk negatively regulates hedgehog signaling by inducing lysosomal degradation of smoothened. BIOCHEM J. 2020;477(1):121–36.
Alman BA. The role of hedgehog signalling in skeletal health and disease. NAT REV RHEUMATOL. 2015;11(9):552–60.
Imuta Y, Nishioka N, Kiyonari H, Sasaki H. Short limbs, cleft palate, and delayed formation of flat proliferative chondrocytes in mice with targeted disruption of a putative protein kinase gene, Pkdcc (AW548124). Dev Dyn. 2009;238(1):210–22.
Kinoshita M, Era T, Jakt LM, Nishikawa S. The novel protein kinase vlk is essential for stromal function of mesenchymal cells. DEVELOPMENT. 2009;136(12):2069–79.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). GENET MED. 2020;22(2):245–57.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. GENET MED. 2015;17(5):405–24.
Barkova E, Mohan U, Chitayat D, Keating S, Toi A, Frank J, Frank R, Tomlinson G, Glanc P. Fetal skeletal dysplasias in a tertiary care center: radiology, pathology, and molecular analysis of 112 cases. CLIN GENET. 2015;87(4):330–7.
Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. AM J MED GENET A. 2019;179(12):2393–419.
Ruano R, Molho M, Roume J, Ville Y. Prenatal diagnosis of fetal skeletal dysplasias by combining two-dimensional and three-dimensional ultrasound and intrauterine three-dimensional helical computer tomography. Ultrasound Obstet Gynecol. 2004;24(2):134–40.
Han J, Yang YD, He Y, Liu WJ, Zhen L, Pan M, Yang X, Zhang VW, Liao C, Li DZ. Rapid prenatal diagnosis of skeletal dysplasia using medical trio exome sequencing: benefit for prenatal counseling and pregnancy management. Prenat Diagn. 2020;40(5):577–84.
Deng L, Liu Y, Yuan M, Meng M, Yang Y, Sun L. Prenatal diagnosis and outcome of fetal hyperechogenic kidneys in the era of antenatal next-generationsequencing. CLIN CHIM ACTA. 2022;528:16–28.
Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, Spiegel E, Brennan K, Stong N, Jobanputra V, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393(10173):758–67.
Yang K, Shen M, Yan Y, Tan Y, Zhang J, Wu J, Yang G, Li S, Wang J, Ren Z, et al. Genetic analysis in fetal skeletal dysplasias by Trio Whole-Exome sequencing. BIOMED RES INT. 2019;2019:2492590.
Plantinga M, Zwienenberg L, van Dijk E, Breet H, Diphoorn J, El MJ, Bouman K, Verheij J, Birnie E, Ranchor AV, et al. Parental experiences of rapid exome sequencing in cases with major ultrasound anomalies during pregnancy. Prenat Diagn. 2022;42(6):762–74.
Galli A, Robay D, Osterwalder M, Bao X, Benazet JD, Tariq M, Paro R, Mackem S, Zeller R. Distinct roles of Hand2 in initiating polarity and posterior shh expression during the onset of mouse limb bud development. PLOS GENET. 2010;6(4):e1000901.
St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072–86.
Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. DEVELOPMENT. 1997;124(1):113–23.
Gao B, Hu J, Stricker S, Cheung M, Ma G, Law KF, Witte F, Briscoe J, Mundlos S, He L, et al. A mutation in Ihh that causes digit abnormalities alters its signalling capacity and range. Nature. 2009;458(7242):1196–200.
Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, Abbott MH, Aughton DJ, Aylsworth AS, Bamshad MJ, et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. AM J HUM GENET. 2005;76(4):609–22.
Probst S, Zeller R, Zuniga A. The hedgehog target Vlk genetically interacts with Gli3 to regulate chondrocyte differentiation during mouse long bone development. DIFFERENTIATION. 2013;85(4–5):121–30.
Acknowledgements
We offer our gratitude to the family for them contribution to this research.
Funding
This research was supported by the Medical and Health Project of Zhejiang Province (Grant No. 2020KY890), Science and Technology Development Program of Ningbo (Grant No. 202002N3150 and 2022S035), and Innovation Project of Distinguished Medical Team in Ningbo (Grant No. 2022020405).
Author information
Authors and Affiliations
Contributions
L.Y. drafted the manuscript; Y.Z. and Y.L. analyzed the WES data, J.C. and J.Z. collected the clinical data; B.L. and D.Z. designed the study; and H.L. made substantial contributions to the interpretation of data. All authors have agreed to be personally accountable for their own contributions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Hospital Ethics Committee of Ningbo Women and Children’s Hospital (approval number EC2020-014). Each patient signed an informed consent prior to enrollment in the study.
Approval and consent statement
The whole methods/steps were implemented according to the related regulations and guidelines.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of the research and any accompanying images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yan, L., Cao, J., Zhang, Y. et al. Prenatal diagnosis to identify compound heterozygous variants in PKDCC that causes rhizomelic limb shortening with dysmorphic features in a fetus from China. BMC Med Genomics 16, 190 (2023). https://doi.org/10.1186/s12920-023-01631-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01631-7