
Abstract
Background
TTN is a complex gene with large genomic size and highly repetitive structure. Pathogenic variants in TTN have been reported to cause a range of skeletal muscle and cardiac disorders. Homozygous or compound heterozygous mutations tend to cause a wide spectrum of phenotypes with congenital or childhood onset. The onset and severity of the features were considered to be correlated with the types and location of the TTN variants.
Methods
Whole-exome sequencing was performed on three unrelated families presenting with fetal akinesia deformation sequence (FADS), mainly characterized by reduced fetal movements and limb contractures. Sanger sequencing was performed to confirm the variants. RT-PCR analysis was performed.
Results
TTN c.38,876–2 A > C, a meta transcript-only variant, with a second pathogenic or likely pathogenic variant in trans, was observed in five affected fetuses from the three families. Sanger sequencing showed that all the fetal variants were inherited from the parents. RT-PCR analysis showed two kinds of abnormal splicing, including intron 199 extension and skipping of 8 bases.
Conclusions
Here we report on three unrelated families presenting with FADS caused by four TTN variants. In addition, our study demonstrates that pathogenic meta transcript-only TTN variant can lead to defects which is recognizable prenatally in a recessive manner.
Similar content being viewed by others


Introduction
Congenital myopathy is a group of diseases with heterogeneous etiology and a wide spectrum of phenotypes [1]. The widespread use of ultrasound screening leads to a high frequency of diagnosis of fetal muscle abnormalities. A diagnosis of severe myopathy always invites considerations regarding termination of pregnancy and inevitably raises concerns about the risk of recurrence in the future pregnancies. Identification of the potential genetic causes facilitates accurate genetic counseling as well as alleviating concerns. The application of next-generation sequencing (NGS) techniques has enabled the genetic basis of neuromuscular diseases to be elucidated, allowing more disease-causing variants to be characterized.
The TTN gene (OMIM: 188,840) encodes titin, a giant sarcomeric protein that plays an important functional and structural role in the sarcomere [2,3,4,5]. Pathogenic variants in TTN were reported to cause a range of skeletal muscle and cardiac disorders, such as dilated cardiomyopathy-1G (CMD1G; OMIM: 604,145), familial hypertrophic cardiomyopathy-9 (CMH9; OMIM: 613,765), tibial muscular dystrophy (TMD; OMIM: 600,334), myofibrillar myopathy-9 with early respiratory failure (MFM9; OMIM: 603,689), and limb-girdle muscular dystrophy type 2 J (LGMD2J; OMIM: 608,807) [6,7,8,9,10]. As one of the most complex human genes, TTN contains 364 exons and four major structural regions (Z disk, I band, A band, and M band), giving rise to a large number of alternatively spliced transcripts [11, 12]. The inferred complete (IC) transcript (NM_001267550.2), also known as meta-transcript, is a theoretical isoform that includes all putative exons (except the exon 48 in transcript NM_133379.5) [13,14,15]. The exons not included in any of the recognized postnatal skeletal muscle isoform (N2A) and cardiac muscle isoforms (N2B, N2BA, Novex-1, Novex-2, and Novex-3) are defined as meta transcript-only exons and are generally thought to be expressed primarily during embryonic development, although some of them have low expression in the postnatal setting [1, 7, 13, 15]. Some studies have proposed that the onset and severity of TTN-related diseases are related to the type and location of the variants [1, 16].
Herein, we report on the investigation of three unrelated families with fetal akinesia deformation sequence (FADS). Three novel TTN variants with a common meta transcript-only variant in trans were identified in our study.
Clinical reports
Family 1
The healthy and non-consanguineous couple has no family histories of neuromuscular diseases or cardiomyopathies. Their first daughter was healthy (Fig. 1A, II-1). During the second pregnancy (Fig. 1A, II-2), the women reported poor fetal movement. Fetal ultrasound in the 22nd week of gestation showed a thickened nuchal fold of 10 mm (normally < 6 mm in the second trimester) and sustained flexed elbows with no joint movements. The pregnancy was terminated in the 24th week after the couple received genetic counsel. Chromosomal karyotype and chromosomal microarray analysis (CMA) on amniotic fluid showed no significant findings.

Pedigree of the three families and segregation of the recessive TTN variants, including the common c.38,876–2 A > C haplotype. (A) Family 1; (B) Family 2; C Family 3
Unfortunately, similar fetal abnormalities were observed during their third pregnancy (Fig. 1A, II-3). Ultrasound screening in the 22nd week of pregnancy revealed abnormal fetal posture with persistent limb joint contractures, rocker-bottom feet, and significantly decreased fetal movement (Fig. 2A-C). The couple terminated the pregnancy in the 23rd week of gestation after genetic counseling.

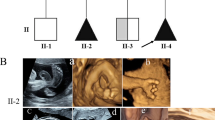
Imaging findings. (A-C) fetal ultrasound images from F1-II0.3. (A) ankle and knee contractures; (B) the left foot presented with rocker bottom deformity; (C) wrist contractures. (D-F) Fetal ultrasound images from F2-II0.2. (D) the abnormal fetal posture with the palms of the feet facing each other; (E) the abnormal curvature of the fetal spine; (F) thickened nuchal fold with cystic the dark area inside. (G-K) Imaging findings from F3-II0.1. (G) flexed position of all limb joints; (H) a right pleural effusion with a width of about 16 mm; (I) mild pericardial effusion with a width of 3.6 mm; (J) widened extracerebral space and mild deformation of the cerebral parenchyma showed by MRI; (K) polyhydramnios with an amniotic fluid index (AFI) of 266 mm and a maximum depth of 95 mm
Family 2
The couple was healthy and non-consanguineous, and their detailed family history was noncontributory. During their first pregnancy (Fig. 1B, II-1), fetal ultrasound in the 25th week of gestation revealed polyhydramnios, hydrops fetalis, scoliosis, flexion of the limbs, and lack of movement. The pregnancy was terminated after genetic counseling. CMA performed on the fetal tissue revealed no pathogenic copy number variants (CNVs). One year later, during the second pregnancy (Fig. 1B, II-2), the tragedy repeated: hydrops fetalis, cystic hygroma, scoliosis, flexion of the limbs, and poor fetal movement were revealed by ultrasonography (US) performed in the 24th week of gestation (Fig. 2D-F). Again, the couple terminated the pregnancy.
Family 3
It was a healthy, non-consanguineous couple, with the age of 28 for the husband and 27 for the wife. This was their first pregnancy (Fig. 1C, II-1). Fetal ultrasound in the 23rd week of gestation indicated contractures of all four limbs, right-sided pleural effusion, and polyhydramnios. All of the fetal abnormalities were confirmed by the following ultrasound in the 31st week (Fig. 2G-I). Pericardial effusion (PEFF) and widened extracerebral space were revealed subsequently by fetal echocardiogram and magnetic resonance imaging (MRI), respectively (Fig. 2J-K). The pregnancy was terminated upon parental request. CMA performed on the fetal tissue didn’t find any pathogenic CNVs.
Methods
WES and data analyses
Genomic DNA was extracted from the muscle tissue of the five aborted fetuses and peripheral blood from the couples. Proband-WES was performed in family C while trio-WES was selected for the other two families. The database of single nucleotide polymorphisms (dbSNP, http://www.ncbi.nlm.nih.gov/snp), the 1000 Genomes Project database (http://browser.1000genomes.org), and genome Aggregation Database (gnomAD v4.0.0, http://gnomad.broadinstitute.org/) was used for searching the minor allele frequencies (MAF < 0.1%) of all known variants. Online bioinformatics tools Mutation Taster (http://www.mutationtaster.org), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), REVEL (https://sites.google.com/site/revelgenomics/), CADD(https://cadd.gs.washington.edu), and SpliceAI (https://github.com/lllumina/SpliceAl) were used to predict the effects of the variants. The pathogenicity of the variants was determined according to the American College of Medical Genetics and Genomics (ACMG) guidelines [17]. Sanger sequencing was performed on family members to confirm segregation and carrier status.
RNA isolation and expression analysis via reverse transcription and PCR
We extracted mRNA from 10 ml of whole blood from the father of family 1 following the manufacturer’s manual (RNAiso Plus, TaKaRa, Japan). Nano Drop 2000 (Thermo Scientific) was used to determine the RNA concentration. The extracted mRNA was synthesized into cDNA (PrimeScriptTM RT reagent Kit, TaKaRa, Japan). PCR was performed using the following primers: 5’-AAAGCCAGAAGCTCCACCTC-3’ (forward primer at the start of exon 192) and 5’-CTCAGGCTCCTCGAACACTT-3’ (reverse primer at the end of exon 205). The cDNA PCR product was visualized by 2% agarose gel electrophoresis and analyzed by Sanger sequencing on the ABI3730xl Genetic Analyzer (Applied Biosystems).
Results
Mutation detection
Heterozygous variant c.38,876–2 A > C was found in all the affected fetuses (Fig. 1). The variant c.38,876–2 A > C, a meta transcript-only variant, is a canonical splicing variant and lies within the proline-glutamine-valine-lysine (PEVK) repeat region (Fig. 3). It was presented in the gnomAD database at a very low frequency of 0.000005611 (9/1,604,030 alleles) in total of the population.

A schematic representation of main titin regions encoded by the inferred complete meta-transcript and the corresponding location of variants identified in the current study
Three truncating variants: c.48396dup (p. Asn16133Ter), c.80,539 C > T (p. Gln26847Ter), and c.15865G > T (p. Glu5289Ter) were found to co-segregate with the c.38,876–2 A > C in the three families, respectively (Fig. 1). Variants c.48396dup (p. Asn16133Ter) and c.80,539 C > T (p. Gln26847Ter) are located in the A-band, while c.15865G > T (p. Glu5289Ter) lies within the I-band (Fig. 3). All three variants are predicted to induce loss of function (LoF) for the encoded titin protein. The frequency of c.48396dup and c.80,539 C > T in the gnomAD v4.0 was 0.000001594 (1/627,338 alleles) and 0.000006580 (1/151,974 alleles), respectively. And c.15865G > T was not recorded in the gnomAD v4.0. According to the ACMG guidelines, c.38,876–2 A > C was classified as likely pathogenic (PM2_Supporting + PVS1_Moderate + PM3_Strong + PP1), c.48396dup was classified as pathogenic (PVS1 + PM2_Supporting + PP1), c.80,539 C > T and c.15865G > T were classified as likely pathogenic (PVS1 + PM2_Supporting). The TTN variants are reported and numbered with exons using the inferred meta-transcript as the reference (NM_001267550.2).
The clinical and molecular features of the three families are summarised in Supplemental Table 1.
RT-PCR and sequencing analysis
To confirm the effect of c.38,876–2 A > C on TTN mRNA, the cDNA from the carrier sample was amplified using primers toward the region between exon 192 and exon 205.
Agarose gel electrophoresis showed that the control sample had only one band with the length of 1042 bp, while the carrier sample showed three bands with lengths of 1034 bp, 1042 bp, and 1151 bp, respectively (Fig. 4a). Sanger sequencing for the abnormal cDNA revealed skipping of 8 nucleotides and retention of intron 199 (Fig. 4b and c). The full length original blot can be seen in Supplemental Fig. 1.

Transcript analyses of TTN variant c.38,876–2 A > C. (A) Display of cropped gels for the RT-PCR fragments, on the right molecular weight (MW) marker. A band of 1042 bp corresponding to the wild-type (WT), a band of 1151 bp corresponding to a retention of intron 199, and a band of 1034 bp corresponding to the skipping of 8 nucleotides. (B) Sanger sequencing of the splicing variant resulting from the retention of the whole intron 199 and the skipping of 8 nucleotides of Exon 200. (C) Schematic representation of the abnormal splicing combining the results of RT-PCR and Sanger sequencing
Discussion
Titin is the largest sarcomeric protein and is organized into four structurally and functionally distinct regions, including Z-disk, I-band, A-band, and M-disk (Fig. 3), each of which has a largely independent functional role [18]. The I-band region is mainly composed of repetitive immunoglobulin (Ig) domains and the PEVK region, which is rich in proline (P), glutamic acid (E), valine (V), and lysine (K), and unravels when stretched to give the muscle elasticity [11, 19]. Exons within the PEVK region are extensively alternatively spliced, regulating passive tension and muscle elasticity.
Previous studies have shown the genotype-phenotype correlations: hereditary myopathy with early respiratory failure (HMERF) is specifically caused by missense variants in the exon 344; tibial muscular dystrophy (TMD) is caused by heterozygous variants in the last exon [20, 21]; all remaining TTN-related skeletal muscle diseases are recessively inherited and present a phenotypic spectrum of severe early-onset disorders collectively termed “congenital titinopathies”, which includes core myopathy with heart disease, centronuclear myopathy, early onset myopathy with fatal cardiomyopathy, and arthrogryposis multiplex congenita [3, 4, 20, 22]. There are several reports of congenital myopathies associated with variants within meta transcript-only exons [1, 3, 7, 13, 14, 23, 24], while variants within meta transcript-only introns is rarely reported [2, 16]. Previous studies showed that the meta transcript-only exons are observably more highly expressed in fetal muscle than adult muscle [2, 15]. It is hypothesized that meta transcript-only variants specifically and selectively affect fetal isoforms, disrupting the development and assembly of fetal muscle, leading to prenatal or congenital severe myopathic phenotypes [3, 15, 16]. In our study, five fetuses with a common meta transcript-only canonical splice site variant all presented with arthrogryposis and a lack of movement, providing a good validation of the above hypothesis.
The c.38,876–2 A > C variant has been previously reported as an incidental finding [25]. And it is not thought to contribute to the fetal phenotypes including skin oedema, pleural effusion, and talipes equinovarus. However, skin oedema and pleural effusion were all observed in our study. We think these are not incidental findings but part of the phenotype. Meanwhile it suggests that characterization of a complete spectrum of TTN-related diseases remains difficult and needs more studies to provide further evidence. Variations in splicing receptor and donor sites are usually considered to lead to abnormal splicing [17]. RNA analysis should be performed since it is a powerful tool to verify the result. It is a great pity that we missed the opportunity to collect muscle tissue from the fetuses. As a remedy, we took blood sample from the carrier for RNA analysis. Despite very low levels of expression in peripheral blood, we succeeded in obtaining the cDNA sequences and confirmed two types of abnormal splicing caused by the mutation. Both abnormal splicing outcomes lead to a frameshift predicted to trigger nonsense mediated decay and result in loss of function (LoF) for the TTN transcript. Since all outcomes are LoF, PVS1 can be applied instead of PVS1_moderate, according to the relevant guidelines [26], and the variant c.38,876–2 A > C is reclassified as pathogenic.
Previous data suggested that variants in exons not expressed at significant levels in cardiac tissue are not associated with cardiomyopathy. Patients with two pathogenic variants predicted to affect both of the cardiac isoforms (N2BA and N2B) have a significantly higher risk of cardiac involvement than those with other combinations of TTN variants [3, 13, 27]. To date, no cardiac phenotype has been reported in individuals with meta transcript-only pathogenic variants [1, 2, 13, 23, 24]. Here, according to a detailed prenatal ultrasound, none of the five fetuses showed signs of cardiac structural malformation.
Heterozygous titin-truncating variants (TTNtv) in exons that are constitutively expressed in cardiac tissue have been identified as the most common genetic cause of dominant or sporadic dilated cardiomyopathy (DCM) [19, 28,29,30,31,32]. As the variants c.48396dup and c.80,539 C > T affect both N2B and N2BA, the two heterozygous carriers (Family 1: I-2 and Family 2: I-2) were referred for cardiac surveillance and the echocardiogram showed normal structural with no sign of cardiomyopathy. The genotype-phenotype correlation for cardiac involvement remains unclear. However, age should not be excluded, as TTNtv-induced cardiomyopathy shows an age-related penetrance [13, 16, 33].
The five fetuses exhibited the major FADS phenotypes such as reduced fetal movements, polyhydramnios, hydrops fetalis, limb contractures, scoliosis, and pleural effusion. One fetus also showed widened extracerebral space which has been reported in very few cases [13], but whether it is caused by TTN mutation is unclear. Although the widespread use of NGS has enabled the discovery of an increasing number of variants in TTN, characterization of a complete spectrum of TTN-related diseases remains difficult due to the high frequency of TTN variants, incomplete penetrance, and as yet unidentified underlying disease mechanisms.
Conclusions
Our finding not only clarify the etiology of the fetal anomaly in the three families, but also help guide their next pregnancy. Pre-implantation genetic diagnosis is a good choice for them. Significant polyhydramnios, arthrogryposis, and reduced fetal movements often indicate neuromuscular disease, and we recommend autosomal recessive congenital titinopathy as one of the differential diagnoses.
Data availability
The raw sequencing data is available in the Sequence Read Archive (SRA) submission: PRJNA1057424.
References
McDermott H, Henderson A, Robinson HK, et al. Novel compound heterozygous TTN variants as a cause of severe neonatal congenital contracture syndrome without cardiac involvement diagnosed with rapid trio exome sequencing. Neuromuscul Disord. 2021;31(8):783–7.
Bryen SJ, Ewans LJ, Pinner J, et al. Recurrent TTN metatranscript-only c.39974-11T > G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat. 2020;41(2):403–11.
Fernández-Marmiesse A, Carrascosa-Romero MC, Alfaro Ponce B, et al. Homozygous truncating mutation in prenatally expressed skeletal isoform of TTN gene results in arthrogryposis multiplex congenita and myopathy without cardiac involvement. Neuromuscul Disord. 2017;27(2):188–92.
Harris E, Töpf A, Vihola A, et al. A ‘second truncation’ in TTN causes early onset recessive muscular dystrophy. Neuromuscul Disord. 2017;27(11):1009–17.
Labeit S, Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270(5234):293–6.
Huang S, Ma Y, Zhang Y, et al. Centronuclear myopathy due to a de novo nonsense variant and a maternally inherited splice-site variant in TTN: a case report. Clin Case Rep. 2021;9(7):e04478.
Cardone N, Moula M, Baelde RJ, et al. Clinical and functional characterization of a long survivor congenital titinopathy patient with a novel metatranscript-only titin variant. Acta Neuropathol Commun. 2023;11(1):48.
Micolonghi C, Fabiani M, Pagannone E, et al. A novel nonsense pathogenic TTN variant identified in a patient with severe dilated cardiomyopathy. Curr Issues Mol Biol. 2023;45(3):2422–30.
Wang G, Lv X, Xu L, et al. Novel compound heterozygous mutations in the TTN gene: elongation and truncation variants causing limb-girdle muscular dystrophy type 2J in a Han Chinese family. Neurol Sci. 2022;43(5):3427–33.
Kellermayer D, Smith JE 3rd, Granzier H. Titin mutations and muscle disease. Pflugers Arch. 2019;471(5):673–82.
Bang ML, Centner T, Fornoff F, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89(11):1065–72.
Freiburg A, Trombitas K, Hell W, et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res. 2000;86(11):1114–21.
Oates EC, Jones KJ, Donkervoort S, et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann Neurol. 2018;83(6):1105–24.
Di Feo MF, Lillback V, Jokela M et al. The crucial role of titin in fetal development: recurrent miscarriages and bone, heart and muscle anomalies characterise the severe end of titinopathies spectrum. J Med Genet. 2023.
Savarese M, Jonson PH, Huovinen S, et al. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet Muscle. 2018;8(1):11.
Savarese M, Vihola A, Oates EC, et al. Genotype-phenotype correlations in recessive titinopathies. Genet Med. 2020;22(12):2029–40.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Schiabor Barrett KM, Cirulli ET, Bolze A, et al. Cardiomyopathy prevalence exceeds 30% in individuals with TTN variants and early atrial fibrillation. Genet Med. 2023;25(4):100012.
Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201–4.
Palmio J, Leonard-Louis S, Sacconi S, et al. Expanding the importance of HMERF titinopathy: new mutations and clinical aspects. J Neurol. 2019;266(3):680–90.
Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71(3):492–500.
Savarese M, Sarparanta J, Vihola A, et al. Increasing role of titin mutations in Neuromuscular disorders. J Neuromuscul Dis. 2016;3(3):293–308.
Alkhunaizi E, Martin N, Jelin AC, et al. Fetal akinesia deformation sequence syndrome associated with recessive TTN variants. Am J Med Genet A. 2023;191(3):760–9.
Chervinsky E, Khayat M, Soltsman S, et al. A homozygous TTN gene variant associated with lethal congenital contracture syndrome. Am J Med Genet A. 2018;176(4):1001–5.
Qin Y, Yao Y, Liu N, et al. Prenatal whole-exome sequencing for fetal structural anomalies: a retrospective analysis of 145 Chinese cases. BMC Med Genomics. 2023;16(1):262.
Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39(11):1517–24.
Ge L, Fu X, Zhang W, et al. Recessive mutations in proximal I-band of TTN gene cause severe congenital multi-minicore disease without cardiac involvement. Neuromuscul Disord. 2019;29(5):350–7.
Loescher CM, Hobbach AJ, Linke WA. Titin (TTN): from molecule to modifications, mechanics, and medical significance. Cardiovasc Res. 2022;118(14):2903–18.
Hinson JT, Chopra A, Nafissi N, et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349(6251):982–6.
Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7(270):270ra6.
Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23(4):980–91.
Romano R, Ghahremani S, Zimmerman T, et al. Reading Frame Repair of TTN truncation variants restores Titin Quantity and functions. Circulation. 2022;145(3):194–205.
Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49(1):46–53.
Acknowledgements
None.
Funding
The work was supported by the Huzhou science and technology program (2022GY10).
Author information
Authors and Affiliations
Contributions
All authors were involved in drafting the article. LF, HL, and MD were involved in the study conception and design. YX and PJ collected the patients’ information. ZL and ML analyzed the ultrasound and MRI results. YQ, XS, YH, YL and GS participated in the experiments and data analysis. MD revised the article critically for important intellectual content. All authors approved the final version to be published.
Corresponding authors
Ethics declarations
Ethical approval
The current investigation was approved by the Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University and the Ethics Committee of Huzhou Maternity & Child Health Care Hospital. All participants provided their written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Fan, L., Li, H., Xu, Y. et al. Identification of four TTN variants in three families with fetal akinesia deformation sequence. BMC Med Genomics 17, 170 (2024). https://doi.org/10.1186/s12920-024-01946-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01946-z