
Abstract
Background
Kagami–Ogata syndrome (KOS) and Temple syndrome (TS) are two imprinting disorders characterized by the absence or reduced expression of maternal or paternal genes in the chromosome 14q32 region, respectively. We present a rare prenatally diagnosed case of recurrent KOS inherited from a mother affected by TS.
Case presentation
The woman’s two affected pregnancies exhibited recurrent manifestations of prenatal overgrowth, polyhydramnios, and omphalocele, as well as a small bell-shaped thorax with coat-hanger ribs postnatally. Prenatal genetic testing using a single-nucleotide polymorphism array detected a 268.2-kb deletion in the chromosome 14q32 imprinted region inherited from the mother, leading to the diagnosis of KOS. Additionally, the woman carried a de novo deletion in the paternal chromosome 14q32 imprinted region and presented with short stature and small hands and feet, indicating a diagnosis of TS.
Conclusions
Given the rarity of KOS as an imprinting disorder, accurate prenatal diagnosis of this rare imprinting disorder depends on two factors: (1) increasing clinician recognition of the clinical phenotype and related genetic mechanism, and (2) emphasizing the importance of imprinted regions in the CMA workflow for laboratory analysis.
Similar content being viewed by others


Background
Genomic imprinting is an epigenetic mechanism in which the expression pattern of a parental allele influences phenotypic expression [1]. Imprinted genes are generally found in clusters and are regulated by imprinting control regions (ICRs), which exhibit parent-specific DNA methylation during germline development. Gamete-specific DNA methylation at ICRs can be retained despite extensive postfertilization epigenetic reprogramming [2, 3].
Kagami–Ogata syndrome (KOS, OMIM #608149) and Temple syndrome (TS, OMIM #616222) are rare imprinting disorders caused by genetic or epigenetic alterations of an imprinted gene cluster in the chromosome 14q32 region. This locus encompasses paternally expressed protein-coding genes (DLK1, RTL1 and DIO3), maternally expressed long noncoding RNAs (MEG3/GTL2, RTL1as and MEG8) and short noncoding RNAs. The absence or reduced expression of maternal or paternal genes results in KOS or TS, respectively [4,5,6].
More than 60% of KOS cases are caused by paternal uniparental disomy 14, approximately 25% are caused by microdeletions, and nearly 10% are caused by epimutations of the chromosome 14q32 imprinted region [7]. The major clinical features of KOS include polyhydramnios, omphalocele, placentomegaly and macrosomia during the prenatal period [4]. KOS, which has a mortality rate of approximately 30%, is also characterized by a small bell-shaped thorax, coat-hanger ribs and a narrow chest wall postnatally, leading to significant respiratory distress upon delivery [8,9,10]. Therefore, prenatal diagnosis is crucial not only for prenatal counseling but also for the management of KOS newborns. However, prenatal identification of KOS is challenging, possibly due to insufficient recognition of this rare disease and its associated imprinted regions.
Here, we present a prenatally diagnosed case of recurrent KOS caused by a deletion in the chromosome 14q32 imprinted region inherited from a mother who was affected by TS. We provide a review of previous cases involving maternal allele deletions associated with KOS, with the objective of improving awareness of rare imprinted disorders within the realm of prenatal diagnosis.
Case presentation
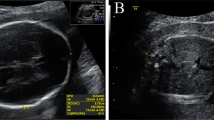
We present the case of a healthy 33-year-old pregnant woman, gravida 3, para 0. She had an ectopic pregnancy, and her second pregnancy ended in stillbirth at 27 weeks gestation, with fetal omphalocele and polyhydramnios indicated by prenatal ultrasound. The miscarried tissues obtained during the second pregnancy were analyzed by single-nucleotide polymorphism (SNP) array (Affymetrix GeneChip 3000Dx, 25 kb 25 marker loss & 25 kb 25 marker gain), and no abnormalities were found. In the third pregnancy, prenatal ultrasound at 23+5weeks of gestation revealed polyhydramnios and clenched hands with overlapping fingers. Additionally, head circumference (226 mm, 92nd percentile for gestational age according to reference data of the Asian population provided by the National Institute of Child Health and Development), abdominal circumference (232.1 mm, 100th percentile for gestational age) and weight (820 g, 99th percentile for gestational age) were above the normal range for a fetus at the corresponding gestational week [11]. The ultrasound abnormalities of the fetus are presented in Fig. 1. Given the woman’s multiple fetal ultrasound abnormalities, gestational age, and adverse pregnancy history, amniocentesis was performed at 27 weeks after the she was provided informed consent. Standard chromosome analyses, SNP array (Affymetrix GeneChip 3000Dx, 25 kb 25 marker loss and 25 kb 25 marker gain) analysis and trio whole exome sequencing (WES) were carried out.

Pedigree of the family and abnormalities of the fetus detected by prenatal ultrasound. A Pedigree of the family: members affected by KOS are colored in black and the member affected by TS is colored in dark gray. ECT, ectopic pregnancy; SB, still birth. B Ultrasound image showing clenched hands with overlapping fingers and polyhydramnios with a deepest vertical pocket of 12.98 cm. B Fetal growth trajectories of head circumference, abdominal circumference and estimated fetal weight, NICHD Fetal Growth Studies—Singletons
Neither aneuploidy nor structural rearrangements of the chromosomes were detected by karyotype analysis. Both SNP array and trio WES revealed a maternal allele deletion of 268.2 kb in the chromosome 14q32 imprinted region, as illustrated in Fig. 2. In light of the abnormalities detected by ultrasound and the results of the genetic tests, KOS was diagnosed prenatally. The diagnosis prompted a review of the SNP array analysis of the previously affected pregnancy, and the same deletion was detected.

SNP array and WES results for the fetus. A A deletion of 268.2 kb at 14q32.2 was detected by SNP array (highlighted in the red frame). B A deletion including DLK1, exons 1–7 of MEG3, IG-DMR and MEG3-DMR was detected by WES (highlighted in a red shadow)
The woman experienced preterm labor at 30 weeks of gestation and delivered a male fetus weighing 2045 g (99th percentile for gestational age) with a head circumference of 30 cm (97th percentile for gestational age), a length of 40 cm (62nd percentile for gestational age), a placental volume of 18 cm × 14 cm × 3 cm and a weight of 500 g [12]. The fetus had facial dysmorphism (small dysplastic ears, small palpebral fissures, full cheeks, tented upper lip, depressed nasal bridge and micrognathia), short neck, thin and slack abdominal wall with abdominal distension, and clenched fist. Chest X-ray revealed a small bell-shaped thorax with distinctive coat-hanger ribs, which are representative features of KOS, as illustrated in Fig. 3 [4]. In addition to feeding difficulties, infections and calcium deficiency, the most prominent symptom of the newborn was respiratory deficiency. The newborn continued to depend on high-frequency oscillatory ventilation with high-quality respiratory support even at one month of age.

Postnatal features of the neonate. A Thin and soft abdominal wall, clenched hands, full cheeks, tented upper lip and micrognathia. B Chest X-ray image showing a typical bell-shaped thorax with coat-hanger ribs
Further clinical evaluations of the mother revealed features of a short stature with a height of 158 cm and small hands and feet. Genetic analysis indicated that the woman carried a de novo deletion in the paternal chromosome 14q32 imprinted region, leading to the diagnosis of TS [13].
Discussion
Herein, we described a case with a prenatal diagnosis of recurrent KOS caused by a 268.2-kb deletion in the chromosome 14q32 imprinted region inherited from the mother who was affected by TS due to a de novo deletion in the paternal chromosome.
Among the three differentially methylated regions (DMRs) in the chromosome 14q32 imprinted region, IG-DMR and MEG3-DMR are hypomethylated, and MEG8-DMR is regulated to be methylated in the maternal chromosome, resulting in the maternal expression of MEG3, RTL1as and MEG8. In contrast, the paternal IG-DMR and the MEG3-DMR are methylated, and the MEG8-DMR is demethylated, regulating the paternal expression of DLK1 and RTL1 [4,5,6]. In our case, the woman carried a de novo deletion in the paternal chromosome. The deleted locus contained DLK1, exons 1-7 of MEG3, IG-DMR and MEG3-DMR. The absence of paternally expressed genes results in TS [4, 13]. When transmitted to the fetus, this deletion was of maternal origin and therefore caused KOS [14]. Preimplantation or prenatal genetic diagnosis was recommended for the next pregnancy due to a recurrence risk of 50%.
KOS is characterized by serial prenatal overgrowth of the fetus and placenta and a narrow chest in the infant. On the other hand, TS is characterized by pre- and postnatal growth restriction, as well as other features, including small hands and feet, short stature, hypotonia, early onset of puberty, and mild dysmorphism of the face. In our case, the recurrent manifestations of two affected pregnancies were consistent with the clinical phenotypes of KOS. The woman had TS features of short stature and small hands and feet. The clinical–genetic diagnoses of TS in the woman and KOS in her offsprings were confirmed by molecular cytogenetics and imprinting mechanisms.
We reviewed 27 previous cases of KOS caused by maternal allele deletions of varying sizes in the literature, as shown in Table 1. Among these cases, there was only one prenatal diagnosis [14], while in most postnatal cases, the diagnosis was performed during infancy and early childhood (20/27 cases). Prenatally, the majority of cases had polyhydramnios (25/27 cases), while omphalocele and placentomegaly were reported in 6 and 5 cases, respectively. Three infants died due to respiratory infection or intracranial hemorrhage [5, 16, 17]. Notably, chromosomal microarray analysis (CMA), including array comparative genomic hybridization (aCGH) and SNP array techniques, detected 18 cases, three of which were attributed to deletions inherited from the mother affected by TS [15, 18, 19]. Furthermore, next-generation sequencing methods such as whole genome sequence (WGS) and copy number variant sequencing (CNV-seq) have also been employed for diagnosing KOS [20, 21].
SNP array-based CMA is routinely utilized as a first-line test in the prenatal diagnosis of fetuses with ultrasound abnormalities [20, 28,29,30]. However, in the application of SNP arrays to detect KOS caused by maternal allele deletions, two important points should be noted. First, copy number variations (CNVs) below the resolution could not be detected. The smallest deletion associated with KOS was only 203 bp, which was far below the detection threshold of the SNP array. However, this deletion was detected by WGS [21]. Second, SNP array analysis workflow primarily focuses on CNVs that contain genes causing human diseases, particularly those dosage-sensitive genes within the OMIM morbid map. In contrast, CNVs that lack OMIM-morbid genes and dosage-sensitive genes may be underestimated. In our case, despite the presence of an imprinting region in the 14q32 deletion CNV, it does not include any OMIM-morbid genes or dosage-sensitive genes. Therefore, the CNV was missed during the CMA-SNP analysis in the second pregnancy. During the analysis in the third pregnancy, recurrent phenotypes prompted a thorough investigation into the imprinting region, subsequently leading to the detection of the CNV.
Currently, some regions associated with imprinting disorders, such as 15q11.2q13 (related to Prader–Willi syndrome and Angelman syndrome), have received ample attention in CMA analysis [31, 32]. However, regions associated with much rarer imprinting disorders, such as KOS, with an estimated incidence of less than 1 in 1 million [33], tend to be underestimated in CMA analysis.
Conclusions
The absence or reduced expression of maternal genes in the 14q32 imprinted region is associated with KOS. Accurate prenatal diagnosis of this rare imprinting disorder depends on two factors: (1) increasing clinician recognition of the clinical phenotype and related genetic mechanism, and (2) emphasizing the importance of imprinted regions in the CMA workflow for laboratory analysts.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- aCGH:
-
Array comparative genomic hybridization
- CMA:
-
Chromosomal microarray analysis
- CNV:
-
Copy number variation
- DMR:
-
Differentially methylated region
- ICR:
-
Imprinting control region
- KOS:
-
Kagami–Ogata syndrome
- OMIM:
-
Online Mendelian Inheritance in Man
- SNP:
-
Single-nucleotide polymorphism
- TS:
-
Temple syndrome
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequence
References
Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC. Genomic imprinting and physiological processes in mammals. Cell. 2019;176:952–65.
Takahashi N, Gray D, Strogantsev R, Noon A, Delahaye C, Skarnes WC, et al. ZFP57 and the targeted maintenance of postfertilization genomic imprints. Cold Spring Harb Symp Quant Biol. 2015;80:177–87.
Takahashi N, Coluccio A, Thorball CW, Planet E, Shi H, Offner S, et al. ZNF445 is a primary regulator of genomic imprinting. Genes Dev. 2019;33:49–54.
Prasasya R, Grotheer KV, Siracusa LD, Bartolomei MS. Temple syndrome and Kagami-Ogata syndrome: clinical presentations, genotypes, models and mechanisms. Hum Mol Genet. 2020;29:R107–16.
Kagami M, O’Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: Hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 2010;6:e1000992.
Beygo J, Mertel C, Kaya S, Gillessen-Kaesbach G, Eggermann T, Horsthemke B, et al. The origin of imprinting defects in temple syndrome and comparison with other imprinting disorders. Epigenetics. 2018;13:822–8.
Li F, Liu S, Jia B, Wu R, Chang Q. Prenatal diagnosis of a mosaic paternal uniparental disomy for chromosome 14: a case report of Kagami-Ogata syndrome. Front Pediatr. 2021;9:691761.
Ogata T, Kagami M. Kagami-Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J Hum Genet. 2016;61:87–94.
Huang H, Mikami Y, Shigematsu K, Uemura N, Shinsaka M, Iwatani A, et al. Kagami-Ogata syndrome in a fetus presenting with polyhydramnios, malformations, and preterm delivery: a case report. J Med Case Rep. 2019;13:340.
Wang X, Pang H, Shah BA, Gu H, Zhang L, Wang H. A male case of Kagami-Ogata syndrome caused by paternal unipaternal disomy 14 as a result of a robertsonian translocation. Front Pediatr. 2020;8:88.
Louis GMB, Grewal J, Albert PS, Sciscione A, Wing DA, Grobman WA, et al. Racial/ethnic standards for fetal growth: the NICHD fetal growth studies. Am J Obstet Gynecol. 2015;213(449):e1–41.
Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr. 2013;13:59.
Gillessen-Kaesbach G, Albrecht B, Eggermann T, Elbracht M, Mitter D, Morlot S, et al. Molecular and clinical studies in 8 patients with temple syndrome. Clin Genet. 2018;93:1179–88.
Jung HS, Vallee SE, Dinulos MB, Tsongalis GJ, Lefferts JA. Maternally inherited 133kb deletion of 14q32 causing Kagami-Ogata syndrome. J Hum Genet. 2018;63:1231–9.
Hu J, Zhang Y, Yang Y, Wang L, Sun Y, Dong M. Case report: Prenatal diagnosis of Kagami-Ogata syndrome in a Chinese family. Front Genet. 2022;13:959666.
Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–42.
Rosenfeld JA, Fox JE, Descartes M, Brewer F, Stroud T, Gorski JL, et al. Clinical features associated with copy number variations of the 14q32 imprinted gene cluster. Am J Med Genet A. 2015;167a:345–53.
Luk HM. Familial Kagami-Ogata syndrome in Chinese. Clin Dysmorphol. 2017;26:124–7.
Sabria-Back J, Monteagudo-Sánchez A, Sánchez-Delgado M, Ferguson-Smith AC, Gómez O, Cartada AP, et al. Preimplantation genetic testing for a chr14q32 microdeletion in a family with Kagami-Ogata syndrome and temple syndrome. J Med Genet. 2022;59:253–61.
Huang R, Yang X, Zhou H, Fu F, Cheng K, Wang Y, et al. Prenatal diagnosis of talipes equinovarus by ultrasound and chromosomal microarray analysis: a Chinese single-center retrospective study. Genes (Basel). 2022;13:1573.
Kilich G, Hassey K, Behrens EM, Falk M, Vanderver A, Rader DJ, et al. Kagami Ogata syndrome: a small deletion refines critical region for imprinting. NPJ Genom Med. 2024;9:5.
Classen CF, Riehmer V, Landwehr C, Kosfeld A, Heilmann S, Scholz C, et al. Dissecting the genotype in syndromic intellectual disability using whole exome sequencing in addition to genome-wide copy number analysis. Hum Genet. 2013;132:825–41.
Beygo J, Elbracht M, De Groot K, Begemann M, Kanber D, Platzer K, et al. Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur J Hum Genet. 2015;23:180–8.
Corsello G, Salzano E, Vecchio D, Antona V, Grasso M, Malacarne M, et al. Paternal uniparental disomy chromosome 14-like syndrome due a maternal de novo 160 kb deletion at the 14q32.2 region not encompassing the IG- and the MEG3-DMRs: patient report and genotype-phenotype correlation. Am J Med Genet A. 2015;167a:3130–8.
Van Der Werf IM, Buiting K, Czeschik C, Reyniers E, Vandeweyer G, Vanhaesebrouck P, et al. Novel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami-Ogata syndrome. Eur J Hum Genet. 2016;24:1724–9.
Altmann J, Horn D, Korinth D, Eggermann T, Henrich W, Verlohren S. Kagami-Ogata syndrome: an important differential diagnosis to Beckwith-Wiedemann syndrome. J Clin Ultrasound. 2020;48:240–3.
Sirera PS, García-Payá E, García JO, Rodríguez RJ, Romero SDH. Maternally inherited deletion encompassing the RTL1as and MEG8 genes of the human 14q32 imprinted region in a patient with a mild Kagami-Ogata syndrome phenotype. Am J Med Genet A. 2023;191:2225–31.
Huang R, Zhou H, Fu F, Li R, Lei T, Li Y, et al. Prenatal diagnosis of Williams-Beuren syndrome by ultrasound and chromosomal microarray analysis. Mol Cytogenet. 2022;15:27.
Hu T, Zhang Z, Wang J, Li Q, Zhu H, Lai Y, et al. Prenatal diagnosis of chromosomal aberrations by chromosomal microarray analysis in fetuses with ultrasound anomalies in the urinary system. Prenat Diagn. 2019;39:1096–106.
Srebniak MI, Boter M, Oudesluijs GO, Cohen-Overbeek T, Govaerts LC, Diderich KE, et al. Genomic SNP array as a gold standard for prenatal diagnosis of foetal ultrasound abnormalities. Mol Cytogenet. 2012;5:14.
Liu S, Zhang K, Song F, Yang Y, Lv Y, Gao M, et al. Uniparental disomy of chromosome 15 in two cases by chromosome microarray: a lesson worth thinking. Cytogenet Genome Res. 2017;152:1–8.
Huang W, Li S, Luo H, Wen X, Lin C, Chen S, et al. Application of various genetic techniques for the diagnosis of Prader-Willi syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2020;37:875–8.
Sakaria RP, Mostafavi R, Miller S, Ward JC, Pivnick EK, Talati AJ. Kagami-Ogata syndrome: case series and review of literature. AJP Rep. 2021;11:e65–75.
Acknowledgements
The authors thank AJE (http://www.aje.cn/) for the English language review.
Funding
This study was supported by the National Key Research and Development Program of China (2022YFC2703302) and the CAMS Innovation Fund for Medical Sciences (CIFMS-2022-I2M-C&T-B-008).
Author information
Authors and Affiliations
Contributions
XTY: analysis and interpretation, data collection, manuscript writing, editing and revision. MML: analysis and interpretation, genetic testing, editing and revision. QWQ: editing and revision. XYZ: editing and revision. NH: editing and revision. YL: study conception and design, analysis and interpretation, data collection, manuscript writing, editing and revision. YLJ: study conception and revision. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics review committee of Peking Union Medical College Hospital (I-23PJ733).
Consent for publication
Written informed consent for publication was obtained from the participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Li, M., Qi, Q. et al. Prenatal diagnosis of recurrent Kagami–Ogata syndrome inherited from a mother affected by Temple syndrome: a case report and literature review. BMC Med Genomics 17, 222 (2024). https://doi.org/10.1186/s12920-024-01987-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01987-4