
Abstract
Purpose
The aim of this study is to delve into the value of N6-Methyladenosine (m6A)-associated genes (MAGs) in pancreatic cancer (PC) prognosis.
Methods
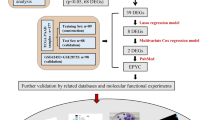
PC sequencing data and corresponding clinicopathological information were retrieved from GEO and TCGA databases. We filtered 19 MAGs in PC specimens and implemented functional annotation in biology. Later, the m6A modification pattern was stratified into m6Acluster A-B according to MAG expression levels, and further categorized into genecluster A-C based on differentially expressed genes between m6Acluster A and B. Next, a MAG-based prognostic prediction model was established by the least absolute shrinkage and selection operator (LASSO) regression analysis and multivariate Cox regression analysis. At last, the role of KRT7 in PC were explored.
Results
We found m6Acluster A pattern presented enrichment pathways associated with cell apoptosis, proliferation, migration, and cancer pathways. Additionally, high-risk group displayed more dismal prognosis and a higher programmed death-ligand 1 expression. The survival prediction ability of the model was verified in three independent PC GEO datasets. KRT7 is the most momentous risk gene in the established prognostic model. Among 18 clinical samples, the KRT7 protein in the surviving patient samples is lower than that in the deceased patient samples. We also identified elevated expression of KRT7 in PC tumor tissues compared to normal tissues using GEPIA 2. Then, the metastasis of PC cells was promoted by overexpressed KRT7 in vitro and in vivo. And IGF2BP3 upregulated KRT7 by increasing the mRNA stability of KRT7.
Conclusions
The PPM built based on CXCL5, LY6K and KRT7 is an encouraging biomarker to define the prognosis. Additionally, IGF2BP3 promoted KRT7 by stabilizing mRNA of KRT7. And KRT7 promoted the metastasis of PC cells by promoting EMT.
Similar content being viewed by others


Background
Pancreatic cancer (PC), deemed as “The King of Carcinoma”, is a commonly encountered malignancy occurring in the digestive tract. As unveiled by the GLOBOCAN 2020 data, the International Agency for Research on Cancer estimates that the number of deaths from PC (466,000) nearly equals to that of cases (496,000) [1]. On a global scale, PC, on account of its dismal prognosis, takes the seventh place among fatal cancers in both men and women, and this ranking is predicted to rise to the second place by 2030 [2]. Among all cancers, PC exhibits the lowest 5-year survival rate (9%) among all types of cancers [3, 4]. Pancreatic adenocarcinoma takes up the largest proportion (85%) in PC [3]. At this stage, useful gene signatures showing efficacy and functioning in prognosis, early detection and improved treatment regimens are still scanty. This phenomenon entails an urgency of developing an approach to predicting the possibility of a prolonged survival and improved prognosis, on the strength of the traits of transcriptome and genome sequencing.
In eukaryotes, N6-methyladenosine (m6A), first reported in the 1970s, is the dominant and abundant type of mRNA modification [5]. m6A is mediated by three categories of individual proteins, namely, writers, erasers, and readers, and their existence may be an indicative of m6A modification. Writers, including METTL 3/14/16, WTAP, VIRMA, ZC3H13 and RBM15/15B, are responsible for the methylation process [6, 7]; erasers, including FTO and ALKBH5, abrogate methylation as demethylases [6, 8]; readers, including YTHDC1/2, HNRNPC, HNRNPA2B1, IGFBP1/2/3, FMR1, LRPPRC, YTHDF1/2/3, IGF2BP3, and RBMX, are methylated effectors, beneficial for the downregulation of corresponding proteins via impairing the stability of target RNAs [6, 9, 10]. The dysregulation of m6A modification displays relevance to carcinogenesis, progression, metastatic spread, and drug resistance of discrepant cancer entities [11, 12]. For example, studies have unravelled that via mRNA m6A modification, METTL14 can inhibit the differentiation of hematopoietic stem/progenitor cells and promote leukemogenesis [13]. Another study has unveiled that IGF2BP3 (a RNA m6A reader) promotes tumor metastasis in triple-negative breast cancer [14]. It has also been demonstrated that WTAP regulates the malignant behaviours of colorectal cancer cells by mediating the m6A of PDK4 [15].
Herein, we delved into the value of m6A-associated genes (MAGs) in PC prognosis, and explored their biological functions in PC, with 19 MAGs as research objects, using public datasets.
Methods
Data sources and clinical specimens
The survival data of MAGs in Figure S1 was downloaded from Gene Expression Profiling Interactive Analysis 2 (GEPIA2, http://gepia2.cancer-pku.cn/#index), with |Log2FC|>0.8, P > 0.05, and matched normal data from TCGA normal and GTEx data as the filtering criteria.
Data pertaining to gene expressions, prognosis and clinicopathological information of PC were downloaded from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) and TCGA (https://portal.gdc.cancer.gov/) databases. We harvested four GEO datasets (GSE21501, GSE28735, GSE62452, and GSE71729) and the TCGA-PAAD dataset for the following research. First, we took the intersection of genes in GSE21501 and TCGA-PAAD, and rectified the merged data with the “sva” package. For transcriptome data, we first perform log transformation on transcriptome data, and then utilize the SVA package ComBat function for batch correction. And we have added this in methods section. From the intersection data, we extracted the expression profiles of 19 MAGs (METTL3, WTAP, ZC3H13, RBM15, RBM15B, YTHDC1/2, HNRNPC, YTHDF1/2/3, FMR1, LRPPRC, HNRNPA2B1, IGFBP2/3, RBMX, FTO, and ALKBH5) in 310 PC patients, as illustrated in Table S1. Later, external validation was achieved by use of GSE28735, GSE62452 and GSE71729 containing 42, 66 and 125 PC specimens with survival information.
A total of 18 clinical specimens used for immunohistochemistry staining were collected in our hospital. All patients have already provided written informed consent.
Consensus clustering analysis of MAGs
A consensus unsupervised clustering analysis was implemented by use of the R package “ConsensusClusterPlus”. According to the MAG expression levels, the m6A pattern was stratified into two discrete subtypes (m6Acluster A-B), and the differences in biological processes between the two modification patterns were clarified on the strength of gene set variation analysis (GSVA) with the hallmark gene set (c2.cp.kegg.symbols and c5.go.symbols) derived from the GSEA/MSigDB database. Adjusted P (adj.P) < 0.05 was indicative of statistically significant difference.
Identification of differentially expressed genes (DEGs) between m6Acluster A and m6Acluster B
The empirical Bayesian approach of “limma” package was applied to determine DEGs between m6Acluster A and m6Acluster B, with logFC > 1 and adj.P < 0.01 as the significance criteria.
Establishment and verification of the MAG-based prognostic prediction model (MAG-PPM)
To build a MAG-PPM based on a scoring system, firstly, a univariate Cox regression analysis was implemented to determine DEGs prominently associated with the prognosis for further assessment (Table S2). Secondly, the m6A modification pattern was further stratified into genecluster A-C for in-depth assessment by use of an unsupervised clustering method as per the expression of prognosis-related DEGs. Finally, a multivariate Cox regression analysis and a least absolute shrinkage and selection operator (LASSO) regression analysis were performed to build the MAG-PPM.
The risk score (RS) was calculated as follows:
RS = Σ (Expi * Coefi).
Where Expi stands for the expression of each gene, and Coefi for the risk coefficient. The gene expression and risk coefficient are shown in Table 1. Based on the median RS, the PC patients fell into high-risk (HR) group and low-risk (LR) group, and the intragroup comparison of the overall survival (OS) rate was conducted via the Kaplan–Meier (KM) survival analysis. Beyond that, we took the measure of the performance of the MAG-PPM in prediction by time-dependent receiver operating characteristic (ROC) curves. Lastly, independent prognostic factors pertaining to OS were clarified via univariate and multivariate Cox regression analysis.
Cell culture
BxPC-3 cells (iCell Bioscience Inc., Shanghai, CHN) were cultured in 90% Dulbecco’s modified Eagle’s medium (SH30243.01, Hyclone, USA) with 10% fetal bovine serum (11011 − 8615, Tianhang, CHN) at 37 °C in 5% CO2 atmosphere.
Cell transfection
SiRNAs targeting KRT7 (KRT7 siRNA) and IGF2BP3 (IGF2BP3 siRNA), KRT7 overexpression vector (KRT7 OE), IGF2BP3 overexpression vector (IGF2BP3 OE) were designed and chemically synthesized by RiboBio (Guangzhou, CHN). Sequences of siRNAs employed herein were listed below: KRT7 siRNA sense: 5’-AAUCUUCUUGUGAUUGUGGGU-3’; anti-sense: 5’-CCACAAUCACAAGAAGAUUCC-3’. IGF2BP3 siRNA sense: 5’- GATATCTCCATTGCAGGAA-3’; anti-sense: 5’-CTTTGTTAGTCCTAAAGAA-3’. siRNA NC: 5’-UUCUCCGAACGUGUCACGUTT-3’. As per the manufacturer’s guideline, Lipofectamine 3000 (Invitrogen, Shanghai, CHN) was employed to transfect BxPC-3 and PANC-1 cells for 24–48 h, after which functional assays of the obtained cells were implemented.
Cell migration and invasion assays
Transwell inserts (Costar, Cambridge, MA) were used to evaluate the potential of cell invasion and migration. 1 × 104 cancer cells were seeded into the coated (for invasion assay) or uncoated (for migration assay) upper chamber with Matrigel (BD Biosciences, San Jose, CA). In the bottom chamber, we filled seven hundred and fifty microlitre complete DMEM medium. We removed the cells in the upper chamber after culturing for 24 h, then with 4% paraformaldehyde fixed the cells in the bottom chamber and with 0.1% crystal violet stained them. Via a light microscope we counted the invaded and migrated cell numbers.
Western blot experiment
We inoculated the cells in six-well plates for four hours, consequently examined the protein expression level of KRT7, E-cadherin, N-cadherin, Vimentin and Snail in the cells. Afterwards, we isolated the total protein from cells via RIPA lysate, and determined its total amount. Afterwards, we taken 100 µg proteins from each group for electrophoresis and transferred them onto PVDF membranes. We used the primary antibody for hypothermal incubation overnight after one hour of sealing at room temperature. With coloring reagent and photographing, we performed color development, then added the secondary antibodies, which were labelled with horseradish peroxide, for one hour of incubation in the next day.
Animals experiment
In this study, two cohorts of animal studies were performed. Control PANC-1 cells (PANC-1vector) or KRT7 overexpression stable PANC-1 cells (PANC-1KRT7) were injected into immunodeficient female mice (n = 3 for each group) through the tail vein. Development of pulmonary metastases was monitored the IVIS@ Lumina II system (Caliper Life Sciences, Hopkinton, MA).
mRNA stability assay
After 48 h of cell transfection, cells were exposed to Act D (Abmole, Shanghai, China) at a concentration of 5 µg/ml for 0 h, 3 h, 6 h, and 9 h. At different time points, the cells were collected and RNA was extracted using Trizol reagent (Invitrogen, Grand Island, NY, USA). After RNA extraction, the KRT7 mRNA level was identified using RT-qPCR as described earlier.
Statistical analysis
R v4.1.3 software and GraphPad Prism v8.0.1 were introduced for statistical processing and visualization. Data were reported as mean ± SD from at least three independent experiments. The data was statistically analysed by two-way ANOVA or Student’s t-test. P < 0.05 was indicative of statistical significance.
Results
Transcriptional alterations of MAGs in PC
Totally 23 MAGs including 8 writers, 2 erasers and 13 readers were extracted from previous literature for this study. First, we explored the expression levels of the 23 MAGs in PC patients by use of GEPIA2 (http://gepia2.cancer-pku.cn/#index). As unveiled in Figure S1, 18 out of 23 MAGs were upregulated in tumour tissues.
Genetic alterations of MAGs in PC
To get a thorough understanding of the expression pattern of MAGs pertaining to tumorigenesis, PC patients from GSE21501 and TCGA-PAAD were integrated for further assays. We took the intersection of genes in GSE21501 and TCGA-PAAD, and rectified the merged data with the “sva” package. From the intersection data, we extracted the expression profiles of 19 MAGs. The expression information of 19 MAGs (METTL3, WTAP, ZC3H13, RBM15/15B, YTHDC1/2, HNRNPC, FMR1, YTHDF1/2/3, LRPPRC, HNRNPA2B1, IGFBP2/3, RBMX, FTO, and ALKBH5) in 310 PC patients is presented in Table S1. The comprehensive landscape of the interactions, correlations and prognostic significance of the 19 MAGs in PC patients is depicted with the MAG network (Fig. 1A). It was observable that the MAG expressions of the same functional category displayed striking interrelations, and prominent interrelations among writers, erasers, and readers were also detectable.
The incidence rates of somatic mutations and copy number variations (CNVs) in 19 MAGs in PC were summarized. Among 173 specimens, only 3.47% experienced mutations in the MAGs (Fig. 1B). Additionally, the CNV frequency test unravelled that CNVs were pervasive in 19 MAGs, dominated by a deletion in copy number (Fig. 1C). Figure 1D illustrates the location of CNVs in the MAGs on chromosomes.

Genetic alterations of m6A-associated genes (MAGs) in pancreatic cancer (PC)
(A) The interaction between MAGs based on GSE21501 and TCGA-PAAD cohort. (B) The mutation frequency of 19 MAGs from TCGA cohort. (C) The CNV variation frequency of MAGs in TCGA cohort. (D) The location of CNV alteration of MAGs using TCGA cohort
m6A modification patterns mediated by 19 MAGs
On the grounds of the expressions of 19 MAGs, patients with qualitatively discrepant m6A modification patterns were categorized by use of the “ConsensusClusterPlus” package, and m6Acluster A and m6Acluster B were ascertained using unsupervised clustering (Fig. 2A). The prognostic assessment of the two discrete patterns unravelled that m6Acluster B was advantageous over m6Acluster A in survival (Fig. 2B), and they exhibited a striking discrepancy in the m6A transcriptional profile (Fig. 2C). Further, GSVA was implemented to ascertain the differences between m6Acluster A differs from m6Acluster B. As unravelled by GO enrichment analysis, pathways in m6Acluster A were related to cell proliferation, migration, aging and differentiation, such as transport of virus, negative regulation of protein tyrosine kinase activity, regulation of cell aging, positive regulation of vascular endothelial cell migration, regulation of erythrocyte differentiation, and hematopoietic stem cell proliferation (Fig. 2D). As demonstrated by the KEGG pathway analysis, pathways in m6Acluster A mainly relevance to cell apoptosis and cancers, such as apoptosis, pathways in cancer, renal cell carcinoma, small cell lung cancer, neurotrophin signalling pathway, and chronic myeloid leukaemia (Fig. 2E).

m6A modification patterns mediated by 19 m6A-associated genes (MAGs)
(A) Differences in clinicopathologic features and expression levels of MAGs between the two distinct subtypes. (B) The prognostic assessment of m6Acluster A and m6Acluster B. (C) PCA analysis showing a remarkable difference in transcriptomes between m6Acluster A and m6Acluster B. (D) GO enrichment analysis between m6Acluster A and m6Acluster B. (E) KEGG enrichment analysis between m6Acluster A and m6Acluster B
Generation of m6A gene signatures
DEGs between m6Acluster A and m6Acluster B were harvested by use of “limma” package, with the aim to further investigate their potential biological behaviours (Figure S2A). Then, the univariate Cox regression unveiled that there were 39 DEGs showing remarkable relevance to the prognosis (Table S2). Further, the m6A patterns were categorized into three genomic subtypes, namely, genecluster A-C, by means of the unsupervised clustering analysis of the 39 DEGs (Figure S2B). It was illustrated by KM curves that patients with genecluster B displayed the optimal OS (Figure S2C). In genecluster A-C, strikingly discrepant expressions of MAGs were observable, identical to the expected results of m6A modification patterns (Figure S2D).
Establishment and verification of the MAG-PPM
The LASSO penalized regression analysis and multivariate Cox regression analysis were implemented on 39 DEGs to evaluate the prognostic performance of the top significant prognostic genes (Fig. 3A and B). A PPM was built on the grounds of KRT7, LY6D, and CXCL5 for PC. The specimens fell into HR and LR groups on the grounds of the threshold of the median RS, where the RS was constructed below: RS = (0.19047896774614 * expression of KRT7) + (0.089224196311407 * expression of LY6D) + (0.113680406489047 * expression of CXCL5). The case distributions in m6Acluster A-B, genecluster A-C, and HS and LS groups are unveiled in Fig. 3C. We found a significant difference in RS between m6Acluster A and m6Acluster B. m6Acluster A showed the lower median score than m6Acluster B (Fig. 3D). Also, the median score reached the minimum in genecluster B but the maximum in genecluster C (Fig. 3E). Additionally, as depicted by the KM survival curves, LS group had a significantly favourable OS compared to that in HS group (Fig. 3F). Beyond that, AUC values of 0.689, 0.670, and 0.618 correspond to the 1-, 3-, and 5-year survival rates of the patients were represented by, respectively (Fig. 3G). Furthermore, PPM was confirmed as an independent prognostic biomarker for evaluating patient outcomes by univariate (Fig. 3H) and multivariate Cox regression analysis (Fig. 3I).

Establishment of the MAG-based prognostic prediction model
(A-B) The LASSO regression analysis and partial likelihood deviance on the prognostic genes. (C) Alluvial diagram of subtype distributions in groups with different risk score and survival outcomes. (D) Differences in risk score among two m6A modification patterns. (E) Differences in risk score among genecluster A-C. (F) Survival analyses for low and high score groups. (G) ROC curves according to the risk score. (H) Forest plot of univariate cox regression analysis. (I) Forest plot of multivariate cox regression analysis
To validate the efficiency of the RS in prognosis, the RS across three external validation groups (GSE28735, GSE62452, GSE71729) was calculated (Fig. 4A-C). The median RS calculated by use of the same formula was taken as the demarcation point, and the patients from the three datasets were divided into HS and LR groups. Consistently, HR group demonstrated worse prognosis than LR group. 1- and 3-year AUC values in GSE28735 were 0.593 and 0.825, respectively (Fig. 4D); 1-, 3-, and 5-year AUC values in GSE62452 were 0.628, 0.879, and 0.911, respectively (Fig. 4E); 1- and 3-year AUC values in GSE71729 were 0.575 and 0.645, respectively (Fig. 4F).
As KRT7 is the most momentous risk gene (Coefi = 0.19) in the established prognostic model, we investigated the underlying mechanism developed by KRT7 expression negatively affects the prognosis of PC. First, 18 PC samples were collected and performed immunohistochemistry. As shown in Fig. 4G, patients with higher expression of KRT7 had poorer survival rates than those with lower expression. Furthermore, we identified elevated expression of KRT7 in PC tumor tissues compared to normal tissues using GEPIA 2 (http://gepia2.cancer-pku.cn/#index), with conditions set at |Log2FC| Cutoff: 1 and p-value Cutoff: 0.001 (Fig. 4H).

verification of the MAG-based prognostic prediction model
(A-C) Validate the prediction effect of RS in GSE28735, GSE62452, and GSE71729 dataset. (D-F) ROC curves to predict the sensitivity and specificity of survival according to the risk score in GSE28735, GSE62452, and GSE71729 dataset. (G) Immunohistochemistry of KRT7 in PC tissues and the relationship between protein levels of KRT7 and prognosis. Bar = 100 μm. (H) We analyze the expression of KRT7 in pancreatic cancer versus normal tissues using the GEPIA 2 database
Inhibition of KRT7 prevents PC cell metastasis
Previous studies have found that KRT7 is closely related to tumor metastasis [16, 17]. And we divided the patients into KRT7 high expression group and low expression group based on the median value of KRT7 mRNA expression. We performed enrichment analysis of DEGs between the KRT7 high expression group and low expression group using FunRich. Results showed that most of the DEGs were enriched in Mesenchymal-to-epithelial transition pathway (Fig. 5A). So, we putdown the KRT7 expression level in BxPC-3 cells by transfecting KRT7 siRNAs. And overexpression the KRT7 expression level in PANC-1 cells (Fig. 5B and C). Then, we performed cell migration and invasion assays. In BxPC-3 cells, cell migration and invasion were inhibited by KRT7 siRNA, and in PANC-1 cells, cell migration and invasion were promoted by KRT7 overexpression (Fig. 5D and E). The epithelial mesenchymal transition related proteins were detected by western blot. The protein level of N-cadherin, Snail, Vimentin were decreased by KRT7 siRNA, and increased by overexpression of KRT7 (Fig. 5F). And the protein level of E-cadherin was increased by KRT7 siRNA, and decreased by overexpression of KRT7 (Fig. 5F).

Inhibition of KRT7 prevents PC cell metastasis
(A) We performed enrichment analysis of DEGs between the high expression group and low expression group of KRT7 using FunRich. (B)The mRNA and (C) protein level of KRT7 in BxPC-3 and PANC-1 cells after transfection. (D) The migration of BxPC-3 and PANC-1 cells after transfection. Bar = 200 μm. (E) The invasion of BxPC-3 and PANC-1 cells after transfection. Bar = 200 μm. (F) The epithelial mesenchymal transition related proteins were detected by western blot. **P < 0.01, ***P < 0.001
KRT7 promoted PC metastasis in vivo
To explore the effects of KRT7 in vivo, KRT7 overexpression stable PANC-1 cells (PANC-1KRT7) and negative control cells (PANC-1vector) were injected into the tail vein of nude mice. Then, we used in vivo imaging system to dynamically monitor the process of lung metastasis (Fig. 6A). Results showed that the PANC-1vector group had significantly fewer lung metastases. And as Fig. 6B and C showed, the PANC-1vector group had significantly fewer lung metastases, compared with the PANC-1KRT7 group.

KRT7 promoted PC metastasis in vivo
Representative bioluminescent images. (B, C) Lungs and H&E-stained lung sections of the mice. Bar = 100 μm. **P < 0.01
IGF2BP3 promoted KRT7 by increasing the mRNA stability of KRT7
To further investigate the molecular mechanism of KRT7, we extracted 23 MAGs from GSE28735 and GSE62452 respectively, and conducted correlation analysis between KRT7 and MAGs. The results showed that the correlation coefficient (0.63 and 0.66) between KRT7 and IGF2BP3 mRNA were the highest (Fig. 7A). Then, we performed western blot assay, and found that IGF2BP3 siRNA decreased the protein level of KRT7 (Fig. 7B). To further determine the mechanism underlying IGF2BP3-induced regulation of KRT7, BxPC-3 and PANC-1 cells were pre-transfected with IGF2BP3 OE or IGF2BP3 siRNA for 24 h. Then we treated cells with the transcription inhibitor actinomycin D (Act D) and found that IGF2BP3 OE enhanced stability of KRT7 mRNA, IGF2BP3 siRNA decreased stability of KRT7 mRNA (Fig. 7C).

IGF2BP3promoted KRT7 by increasing the mRNA stability of KRT7
(A) Correlations between KRT7 and MAGs in GSE62452 and GSE28735. (B) Western blot assay was performed to detect the KRT7 protein expression. (C) The mRNA stability of KRT7 was checked by qRT-PCR
Evaluation of TME and checkpoints between HR and LR groups
How RS and immune cell interrelate was estimated by use of the CIBERSORT algorithm. It was unveiled in Figure S3A, RS exhibited positive relevance to macrophages and neutrophils and negative relevance to naive B cells, monocytes, and CD8+ T cells. By the same token, we delved into how KRT7, LY6D, and CXCL5 relate to immune cell abundance. Observably, there were associations between several immune cells and the genes (Figure S3B). Beyond that, we found that PD-L1 was highly expressed in HS group (Figure S3C).
Discussion
PC is a lethal disease with a high death rate, and current PC cases present an annual upward trend [18], so it comes into the spotlight of relevant research. Most patients have developed into the locally advanced or metastatic stage when diagnosed, due to the lack of early and effective screening. As such, PC cases have a high propensity for relapse and death in spite of the condition that they have been surgically treated and received adjuvant chemotherapy or other standardized managements [19, 20]. This entails an urgency to search for applicable biomarkers for early PC screening and OS prediction for PC cases.
With the development of sequencing technology, relying on the big data platform and using the bioinformatics principle to explore the factors affecting the development of disease at gene level are adopted by more and more researchers [21,22,23,24]. In present study, we harvested four GEO datasets (GSE21501, GSE28735, GSE62452, and GSE71729) and the TCGA-PAAD dataset. We merged GSE21501 and TCGA-PAAD datasets for research, and external validation by use of GSE28735, GSE62452 and GSE71729 datasets.
Here, we revealed two distinct m6A modification patterns (m6Acluster A and B) based on 19 MAGs. The OS turned out to be more unfavourable in patients with m6Acluster A relative to those with m6Acluster B. Then, genecluster A-C were identified obtained on the grounds of the DEGs between m6Acluster A and B. We found that the expression level of most of 19 MAGs were lower in m6Acluster B cases than in m6Acluster A cases, and m6Acluster B cases had better OS. The expression level of DEGs were lower in genecluster C, and genecluster C cases had better OS. Further, the PPM was built on the grounds of KRT7, LY6D, CXCL5. KRT7 has previously validated to be overexpressed in numerous cancers and associated with unsatisfactory survival outcomes in cervical cancer, breast cancer, ovarian cancer, etc [25,26,27]. CXCL5 expression is raised in thyroid cancer cells and tissues. The results of in vitro experiments unravelled that the downregulated CXCL5 strikingly attenuated the malignant behaviour of tumor cells [28]. CXCL5 overexpression promotes tumour development by promoting epithelial-mesenchymal transition (EMT) [29]. CXCL5 knockdown repressed the proliferation, migration, and expression of EMT indexes of PC cells in vitro, and curbed the growth of xenograft tumours in vivo as well [30]. LY6D is upregulated in laryngeal cancer and may serve as a biomarker for chemoresistance in cancer stem cells [31]. Tethering LY6D expression helps blockade cancer cell growth and metastasis [16, 17]. A higher LY6D expression level heralds a higher aggression and worse outcomes of PC [32].
The specimens fell into HS and LS groups as per the threshold of the median RS. Integrated analyses yielded a prominently shorter OS in HS group relative to LS group, and ascertained that RS of PPM is the independent diagnostic biomarker in PC. Further, the credible efficacy of the RS prognostic model was ascertained by three external validation datasets (GSE28735, GSE62452, GSE71729) and ROC analysis. Among 18 clinical samples, the KRT7 protein in the surviving patient samples was lower than that in the deceased patient samples. And by using GEPIA 2, we found that KRT7 was upregulated in PC relative to normal tissues. Moreover, we divided the patients into KRT7 high expression group and low expression group based on the median value of KRT7 mRNA expression. We performed enrichment analysis of DEGs between the KRT7 high expression group and low expression group using FunRich, and found that most of the DEGs were enriched in Mesenchymal-to-epithelial transition pathway. In vitro and in vivo experiments also determined that overexpressed KRT7 promoted the metastasis of PC cells. To further investigate the molecular mechanism of KRT7, we conducted the correlation analysis between KRT7 and 23 MAGs in GSE62452 and GSE28735, respectively. The results showed that the correlation coefficient between KRT7 and IGF2BP3 mRNA was the highest. Most importantly, IGF2BP3 has been identified as an m6A reader, promoting the expression of HIF1A by enhancing RNA stability [33]. Then, by western blot assay and mRNA stability assay, we found that IGF2BP3 enhanced the mRNA stability of KRT7 to regulate KRT7 expression.
However, this research has several limitations. Firstly, the model we constructed is based solely on public databases, and more clinical sample data and an independent validation cohort are needed to validate its effectiveness. Secondly, key genes in the model include CXCL5 and LY6K, and their functions and mechanisms in PC require further research. Lastly, even we found that IGF2BP3 can regulate the expression of KRT7 and the mRNA stability of KRT7, further exploration of the molecular mechanism is still needed.
Conclusions
Altogether, the PPM built based on CXCL5, LY6K and KRT7 performs well in survival prediction. And IGF2BP3 enhanced the mRNA stability of KRT7 to regulate KRT7 expression. and overexpressed KRT7 promoted the metastasis of PC cells in vitro and in vivo.
Data availability
The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Abbreviations
- m6A:
-
N6-Methyladenosine
- MAG :
-
m6A-associated gene
- PC:
-
pancreatic cancer
- LASSO:
-
least absolute shrinkage and selection operator
- GEPIA2:
-
Gene Expression Profiling Interactive Analysis 2
- GEO:
-
Gene Expression Omnibus
- GSVA:
-
gene set variation analysis
- DEGs:
-
differentially expressed genes
- PPM:
-
prognostic prediction model
- KRT7 OE:
-
KRT7 overexpression vector
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Li F, Zhang P. The N6-Methyladenosine- (m6A-) Associated Genes Act as Strong Key Biomarkers for the Prognosis of Pancreatic Adenocarcinoma. Comput Math Methods Med 2021, 2021:8715823.
Wang L, Zhang S, Li H, Xu Y, Wu Q, Shen J, Li T, Xu Y. Quantification of m6A RNA methylation modulators pattern was a potential biomarker for prognosis and associated with tumor immune microenvironment of pancreatic adenocarcinoma. BMC Cancer. 2021;21(1):876.
Lin B, Pan Y, Yu D, Dai S, Sun H, Chen S, Zhang J, Xiang Y, Huang C. Screening and Identifying m6A Regulators as an Independent Prognostic Biomarker in Pancreatic Cancer Based on The Cancer Genome Atlas Database. Biomed Res Int 2021, 2021:5573628.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971–5.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27(5):626–41.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. M(6)a RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014;8(1):284–96.
Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15(5):313–26.
Fang Z, Mei W, Qu C, Lu J, Shang L, Cao F, Li F. Role of m6A writers, erasers and readers in cancer. Exp Hematol Oncol. 2022;11(1):45.
Huang Q, Mo J, Liao Z, Chen X, Zhang B. The RNA m(6)a writer WTAP in diseases: structure, roles, and mechanisms. Cell Death Dis. 2022;13(10):852.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. METTL14 inhibits hematopoietic Stem/Progenitor differentiation and promotes Leukemogenesis via mRNA m(6)a modification. Cell Stem Cell. 2018;22(2):191–205. e199.
Jiang T, He X, Zhao Z, Zhang X, Wang T, Jia L. RNA m6A reader IGF2BP3 promotes metastasis of triple-negative breast cancer via SLIT2 repression. FASEB J. 2022;36(11):e22618.
Dai X, Chen K, Xie Y. WTAP mediated the N6-methyladenosine modification of PDK4 to regulate the malignant behaviors of colorectal cancer cells in vitro and in vivo. Curr Med Chem 2022.
An Q, Liu T, Wang MY, Yang YJ, Zhang ZD, Liu ZJ, Yang B. KRT7 promotes epithelial–mesenchymal transition in ovarian cancer via the TGF–beta/Smad2/3 signaling pathway. Oncol Rep. 2021;45(2):481–92.
Chen F, Chen Z, Guan T, Zhou Y, Ge L, Zhang H, Wu Y, Jiang GM, He W, Li J, et al. N(6) -Methyladenosine regulates mRNA Stability and Translation Efficiency of KRT7 to promote breast Cancer lung metastasis. Cancer Res. 2021;81(11):2847–60.
Guo Z, Zhang X, Lin C, Huang Y, Zhong Y, Guo H, Zheng Z, Weng S. METTL3-IGF2BP3-axis mediates the proliferation and migration of pancreatic cancer by regulating spermine synthase m6A modification. Front Oncol. 2022;12:962204.
Song Z, Wang X, Chen F, Chen Q, Liu W, Yang X, Zhu X, Liu X, Wang P. LncRNA MALAT1 regulates METTL3-mediated PD-L1 expression and immune infiltrates in pancreatic cancer. Front Oncol. 2022;12:1004212.
Neoptolemos JP, Springfeld C, Hackert T. A review of pancreatic Cancer. JAMA. 2021;326(23):2436.
Akacin I, Ersoy S, Doluca O, Gungormusler M. Comparing the significance of the utilization of next generation and third generation sequencing technologies in microbial metagenomics. Microbiol Res. 2022;264:127154.
Mannarapu M, Dariya B, Bandapalli OR. Application of single-cell sequencing technologies in pancreatic cancer. Mol Cell Biochem. 2021;476(6):2429–37.
Su Y, Qi R, Li L, Wang X, Li S, Zhao X, Hou R, Ma W, Liu D, Zheng J, et al. An immune-related gene prognostic risk index for pancreatic adenocarcinoma. Front Immunol. 2022;13:945878.
Wang S, Hu Y, Lv X, Li B, Gu D, Li Y, Sun Y, Su Y. Circ-0000284 arouses malignant phenotype of cholangiocarcinoma cells and regulates the biological functions of peripheral cells through cellular communication. Clin Sci (Lond). 2019;133(18):1935–53.
Benti S, Tiwari PB, Goodlett DW, Daneshian L, Kern GB, Smith MD, Uren A, Chruszcz M, Shimizu LS, Upadhyay G. Small molecule binds with lymphocyte Antigen 6K to Induce Cancer Cell Death. Cancers (Basel) 2020, 12(2).
Mayama A, Takagi K, Suzuki H, Sato A, Onodera Y, Miki Y, Sakurai M, Watanabe T, Sakamoto K, Yoshida R, et al. OLFM4, LY6D and S100A7 as potent markers for distant metastasis in estrogen receptor-positive breast carcinoma. Cancer Sci. 2018;109(10):3350–9.
Song J, Ruze R, Chen Y, Xu R, Yin X, Wang C, Xu Q, Zhao Y. Construction of a novel model based on cell-in-cell-related genes and validation of KRT7 as a biomarker for predicting survival and immune microenvironment in pancreatic cancer. BMC Cancer. 2022;22(1):894.
Chang W, Chang Q, Lu H, Liu S, Li Y, Chen C. MicroRNA-873-5p suppresses cell malignant behaviors of thyroid cancer via targeting CXCL5 and regulating P53 pathway. Hum Vaccin Immunother. 2022;18(5):2065837.
Ge X, He J, Wang L, Zhao L, Wang Y, Wu G, Liu W, Shu Y, Gong W, Ma XL, et al. Epigenetic alterations of CXCL5 in cr(VI)-induced carcinogenesis. Sci Total Environ. 2022;838(Pt 1):155713.
Wang ZZ, Li XT, Li QJ, Zhou JX. Targeting CXCL5 in pancreatic Cancer cells inhibits Cancer Xenograft Growth by reducing proliferation and inhibiting EMT progression. Dig Dis Sci 2022.
Wang J, Fan J, Gao W, Wu Y, Zhao Q, Chen B, Ding Y, Wen S, Nan X, Wang B. LY6D as a Chemoresistance Marker Gene and therapeutic target for laryngeal squamous cell carcinoma. Stem Cells Dev. 2020;29(12):774–85.
Barros-Silva JD, Linn DE, Steiner I, Guo G, Ali A, Pakula H, Ashton G, Peset I, Brown M, Clarke NW, et al. Single-cell analysis identifies LY6D as a marker linking castration-resistant prostate luminal cells to prostate progenitors and Cancer. Cell Rep. 2018;25(12):3504–e35183506.
Cheng K, Liu S, Li C, Zhao Y, Wang Q. IGF2BP3/HIF1A/YAP signaling plays a role in driving acute-on-chronic liver failure through activating hepatocyte reprogramming. Cell Signal. 2023;108:110727.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
XC designed this work, performed experiments, and wrote this manuscript. SW and YS integrated and analyzed the data. All authors revised and approved this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This work was approved by the Academic Committee of The Affiliated Huaian No.1 People’s Hospital of Nanjing Medical University and was conducted following the Declaration of Helsinki and the institutional guidelines for the use of laboratory animals.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Table S1:
The expression profiles of 19 m6A-associated genes in pancreatic cancer patients collected from GSE21501 and TCGA-PAAD datasets.
Supplementary Material 2: Figure. S1:
Transcriptional alterations of m6A-associated genes (MAGs) in pancreatic cancer (PC)
The expression levels of the 23 MAGs in PC patients by use of GEPIA2 (http://gepia2.cancer-pku.cn/#index)
Supplementary Material 3: Figure. S2:
Generation of m6A gene signatures
(A) There are 76 DEGs between m6Acluster A and m6Acluster B. (B) Differences in clinicopathologic features and expression levels of DEGs between the three distinct subtypes. (C) The prognostic assessment of genecluster A-C. (D) The expression of m6A-associated genes among the genecluster A-C.
Supplementary Material 4: Figure. S3:
Evaluation of TME and checkpoints between high and low risk groups
(A) Correlations between risk score and immune cell types. (B) Correlations between the abundance of immune cells and three genes in the MAG-based prognostic prediction model. (C) The expression of PD-L1 in high and low risk groups.
Supplementary Material 5: Table S2:
39 differentially expressed genes between m6Acluster A and m6Acluster B were remarkable relevance to the prognosis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, C., Wang, S. & Sun, Y. The role of KRT7 in metastasis and prognosis of pancreatic cancer. Cancer Cell Int 24, 321 (2024). https://doi.org/10.1186/s12935-024-03500-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03500-4