
Abstract
Programmed death receptor-1 (PD-1) and its ligand, programmed death ligand-1 (PD-L1) are essential molecules that are key in modulating immune responses. PD-L1 is constitutively expressed on various immune cells, epithelial cells, and cancer cells, where it functions as a co-stimulatory molecule capable of impairing T-cell mediated immune responses. Upon binding to PD-1 on activated T-cells, the PD-1/PD-L1 interaction triggers signaling pathways that can induce T-cell apoptosis or anergy, thereby facilitating the immune escape of tumors. In urological cancers, including bladder cancer (BCa), renal cell carcinoma (RCC), and prostate cancer (PCa), the upregulation of PD-L1 has been demonstrated. It is linked to poor prognosis and enhanced tumor immune evasion. Recent studies have highlighted the significant role of the PD-1/PD-L1 axis in the immune escape mechanisms of urological cancers. The interaction between PD-L1 and PD-1 on T-cells further contributes to immunosuppression by inhibiting T-cell activation and proliferation. Clinical applications of PD-1/PD-L1 checkpoint inhibitors have shown promising efficacy in treating advanced urological cancers, significantly improving patient outcomes. However, resistance to these therapies, either intrinsic or acquired, remains a significant challenge. This review aims to provide a comprehensive overview of the role of the PD-1/PD-L1 signaling pathway in urological cancers. We summarize the regulatory mechanism underlying PD-1 and PD-L1 expression and activity, including genetic, epigenetic, post-transcriptional, and post-translational modifications. Additionally, we discuss current clinical research on PD-1/PD-L1 inhibitors, their therapeutic potential, and the challenges associated with resistance. Understanding these mechanisms is crucial for developing new strategies to overcome therapeutic limitations and enhance the efficacy of cancer immunotherapy.
Graphic abstract

Similar content being viewed by others

Introduction
Cancers of the urinary system, such as bladder cancer (BCa), renal cell carcinoma (RCC), and prostate cancer (PCa), pose substantial challenges in the field of oncology. BCa, which is mainly transitional cell carcinoma, occurs more frequently in men and can be classified by the degree of invasion into non-muscle invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC) [1,2,3]. The standard treatment for NMIBC usually includes transurethral resection of the bladder tumor (TURBT) along with localized therapies to reduce the risk of recurrence [4, 5]. However, NMIBC recurs locally in 60–70% of cases, with 15–40% progressing to MIBC or distant metastasis [6, 7]. Epidemiological data indicate that the incidence of BCa varies globally, with higher rates typically observed in developed countries. The primary risk factors include smoking, occupational exposure, and chronic inflammation [8]. Smoking is considered the most significant modifiable risk factor, with studies showing that smokers are more than three times as likely to develop BCa compared to non-smokers [9]. Additionally, occupational exposure to certain chemicals, such as aniline dyes, chlorides, and chemicals related to aluminum production, significantly increases the risk of developing BCa [2]. Long-term chronic bladder inflammation, urinary tract infections, and certain parasitic infections, such as schistosomiasis, are also closely associated with the development of BCa. In terms of diagnosis, cystoscopy is regarded as the gold standard for BCa diagnosis. Cystoscopy allows for direct visualization of the bladder mucosa, enabling early detection of tumor lesions and the possibility of performing biopsies to obtain pathological evidence. However, the sensitivity and specificity of cystoscopy may be limited by the size and location of the tumor. Therefore, imaging techniques play a crucial role in the diagnosis and staging of BCa. Computed tomography (CT) and magnetic resonance imaging (MRI) are particularly essential in assessing tumor invasion depth, lymph node involvement, and distant metastasis. In recent years, molecular testing techniques have increasingly been applied to the diagnosis and characterization of BCa. Ki-67, TP53, and CK20 are key biomarkers for diagnosing and predicting BCa outcomes [10]. Ki-67 indicates tumor cell growth, with higher levels suggesting more aggressive tumors and poorer prognosis. TP53 often mutates in BCa, contributing to tumor progression and therapy resistance. CK20 can identify BCa subtypes, confirming urothelial origin and providing insights into tumor differentiation and invasiveness. Besides, FGFR3 and TERT promoter mutations have been identified as key molecular markers associated with BCa [11, 12]. These markers not only aid in the early diagnosis of tumors but also provide a basis for personalized treatment strategies. Surgery, chemotherapy, immunotherapy, and targeted therapies are essential for BCa treatment. For localized BCa, transurethral resection of bladder tumors (TURBT) and radical cystectomy are primary surgical treatments that effectively control the disease [13]. However, for advanced or metastatic cases, surgery alone is insufficient, making chemotherapy the preferred supplement. Cisplatin-based regimens like MVAC and GC are commonly used and effective. Recently, PD-1/PD-L1 immune checkpoint inhibitors have shown significant success in advanced BCa, providing an alternative for patients who cannot undergo chemotherapy [14]. Additionally, targeted therapies, such as those addressing FGFR3 mutations, offer personalized options for certain bladder cancer subtypes. Combining these treatments can improve survival rates and quality of life for bladder cancer patients. However, the 5-year survival rate remains relatively low at 20–40%, with around 50% of patients developing distant metastases within 3 years and a mere 10% surviving up to 5 years [7, 15]. RCC, which accounts for 2–3% of all cancers, originates from the renal tubular epithelium. The most common subtype is clear cell renal cell carcinoma (ccRCC) [16]. RCC is frequently asymptomatic in its early stages and is often diagnosed incidentally. At the time of diagnosis, up to 30% of cases have already metastasized [17, 18]. Despite surgical resection, about 30% of organ-confined cases experience metastasis during follow-up [19, 20]. The prognosis for advanced or metastatic RCC remains bleak, with a less than 10% survival rate at 5 years post-diagnosis, compounded by resistance to conventional therapies like chemotherapy and radiotherapy [18, 21]. PCa, prevalent in the male genitourinary system, demonstrates a promising 10-year overall survival rate of 99% for early-stage localized cases due to enhanced detection and treatment strategies [22, 23]. However, up to 25% of patients experience biochemical recurrence after initial treatment, progressing to metastatic castration-resistant prostate cancer (mCRPC) [24,25,26]. Once advanced, the median survival period drops to 13–32 months, with a 5-year survival rate of only 15% [27, 28]. Effective management strategies for urinary system tumors remain crucial given their profound impact on patient outcomes and societal health.
Programmed death receptor-1 (PD-1) and its ligand, programmed death ligand-1 (PD-L1), are essential immune checkpoint molecules that play a pivotal role in the evasion of tumor immune responses [29]. PD-1 is a transmembrane receptor predominantly located on activated T cells, B cells, NK cells, and monocytes [30], The ligand PD-L1, primarily found on the surfaces of tumor cells, interacts with its receptor PD-1 [31]. This interaction results in the suppression of T cells, hindering their ability to detect and destroy tumor cells, thus aiding immune evasion. Inhibitors that target the PD-1/PD-L1 pathway disrupt this interaction, thereby restoring the cytotoxic functions of T cells to efficiently attack and eliminate tumors, countering immune evasion [32]. Additionally, PD-L1 can be expressed by various cells within the tumor microenvironment (TME) [33, 34], creating an immunosuppressive milieu that further supports tumor progression [35, 36]. These cells not only suppress T cell activity via the PD-1/PD-L1 pathway but also secrete immunosuppressive cytokines, exacerbating immune evasion [37]. Additionally, conditions like hypoxia, elevated lactate levels, and nutrient deficiencies in the tumor microenvironment can lead to PD-L1 expression, thereby increasing the tumor’s ability to avoid immune detection [38]. In recent years, immunotherapies targeting the PD-1/PD-L1 axis have made substantial progress [39,40,41]. Inhibitors such as Nivolumab and Pembrolizumab for PD-1, as well as Atezolizumab and Durvalumab for PD-L1, have shown notable advancements, and have demonstrated notable efficacy across diverse cancer types. These drugs function by disrupting PD-1/PD-L1 interactions, releasing T cells from suppression, restoring their anti-tumor activity, and thereby achieving therapeutic efficacy.
Anti-PD-L1 and PD-1 therapies, widely used and recommended by European Society for Medical Oncology (ESMO) and the National Comprehensive Cancer Network (NCCN), are effective for urological cancers [42,43,44,45,46,47]. Atezolizumab and pembrolizumab, in particular, show significant results in advanced or metastatic bladder cancer, renal cell carcinoma, and prostate cancer. These therapies are suggested as first or second-line treatments, especially for patients who have failed chemotherapy or are ineligible for it, and have become essential in standard treatment regimens, greatly improving patient outcomes. This review seeks to establish a foundational understanding of the immune system’s structure and function, with a particular focus on the PD-1/PD-L1 axis. It summarizes key clinical trials that have resulted in the approval of the five PD-1/PD-L1 inhibitors currently used to treat urological cancers. Finally, the review addresses current and future challenges in the application of PD-1 and PD-L1 inhibitors, highlighting the essential role of predictive biomarkers.
PD-1/PD-L1 pathway: molecular and cellular mechanisms
Structure and function of PD-1 and PD-L1
PD-1 is a suppressive receptor found on the surface of immune cells such as T cells, B cells, and NK cells [48]. As a member of the immunoglobulin superfamily, PD-1 features an IgV-like extracellular domain, a transmembrane segment, and an intracellular region. The intracellular part includes an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM), both essential for PD-1 signaling [49]. Likewise, PD-L1, also part of the immunoglobulin superfamily, possesses an extracellular domain with IgV-like and IgC-like structures, a transmembrane component, and a cytoplasmic tail [50]. PD-L1 is broadly expressed in various cell types, including antigen-presenting cells (APCs), tumor cells, and certain non-immune cells [51]. The PD-1/PD-L1 pathway mainly dampens T cell activation and effector functions, ensuring immune system equilibrium [52]. The interaction between PD-1 and PD-L1 triggers downstream signaling pathways, particularly via the phosphatase SHP-2 containing Src homology 2 domain, which inhibits T cell receptor (TCR) and CD28 co-stimulatory signals [39, 53]. This results in suppressed T cell proliferation, cytokine production, and cytotoxic activity, potentially leading to T cell apoptosis or anergy [54, 55]. While this mechanism typically prevents autoimmune responses, tumor cells exploit it within the tumor microenvironment (TME) to escape immune detection.
Regulation of PD-1/PD-L1 expression
The expression of PD-1 and PD-L1 is influenced by a range of genetic and epigenetic elements. The promoter region of PD-1 has numerous sites for transcription factor binding, including nuclear factor kappa B (NF-κB), activator protein 1 (AP-1), and interferon regulatory factors (IRF), among others [56, 57]. These transcription factors can enhance PD-1 gene transcription under different stimulatory conditions. DNA methylation and histone modifications also critically influence PD-1/PD-L1 expression [58, 59]. High methylation of the PD-1 promoter is associated with low expression, whereas demethylation promotes its expression. Additionally, inhibition of histone deacetylases (HDACs) can increase PD-1 expression [60, 61]. The expression of PD-L1 is controlled by various signaling pathways, including those involving interferon-gamma (IFN-γ) and transforming growth factor-beta (TGF-β) [62, 63]. IFN-γ directly enhances PD-L1 transcription by activating IRF-1 [64]. Additionally, certain microRNAs (miRNAs) play a role in modulating PD-L1 levels [65]. Elements within the tumor microenvironment (TME) also trigger PD-L1 expression, aiding tumor cells in evading the immune response [66, 67]. Hypoxia significantly influences PD-L1 regulation through hypoxia-inducible factor 1-alpha (HIF-1α), which stabilizes and increases PD-L1 gene expression under low oxygen conditions [68]. Lactic acid, another key component of the TME, upregulates PD-L1 via the STAT3 signaling pathway [69]. Furthermore, cytokines such as IFN-γ, tumor necrosis factor-alpha (TNF-α), and interleukin-10 (IL-10) markedly boost PD-L1 expression levels [70, 71], typically secreted by immune cells infiltrating the tumor, such as T cells, macrophages, and dendritic cells (DCs) [72]. The model of PD-L1 and PD-1 interaction is displayed in Fig. 1.

The model of PD-L1 and PD-1 interaction. T cell apoptosis is initiated when PD-L1 binds to its receptor PD-1, leading to T cell exhaustion and immune evasion. PD-1/PD-L1 antibodies disrupt the interaction between PD-1 and PD-L1, enabling T lymphocytes to reactivate, proliferate, and target tumor cells for destruction. PD-L1 expression is upregulated in response to inflammatory signals such as IFN-γ produced by activated T cells
PD-1/PD-L1 signaling in cancer and immune cells
PD-L1 is prominently upregulated in various cancer types, including urogenital cancers, where its high expression on tumor cells binds to PD-1 on T cells, effectively inhibiting T cell anti-tumor activity and promoting tumor growth and progression [73]. Furthermore, PD-L1 can be released via exosomes, further enhancing its inhibitory effects by engaging PD-1 [74]. This mechanism of immune evasion allows tumors to evade host immune surveillance and expand. PD-L1 is not confined to tumor cells but can also be expressed in stromal cells surrounding the tumor, which induce PD-L1 expression through the secretion of cytokines and growth factors [75]. Additionally, hypoxic conditions in the TME regulate PD-L1 expression through upregulation of HIF-1α, adding complexity to treatment strategies [76]. Beyond its role in tumor cells, the PD-1/PD-L1 signaling pathway plays a pivotal role in immune cell interactions [77]. In APCs like DCs and macrophages, PD-L1 expression regulates their functions, influencing antigen presentation and cytokine secretion [78]. PD-L1 inhibits DC maturation and function, thereby reducing their capacity to activate T cells and suppressing anti-tumor immune responses [79]. Additionally, the PD-1/PD-L1 pathway is critical in regulatory T cells (Tregs), enhancing their suppressive functions. PD-1 expression in Tregs helps maintain immune tolerance by secreting inhibitory cytokines and directly suppressing effector T cells (Teffs), thereby preventing excessive immune responses and autoimmune diseases [80]. In the TME, Tregs’ functions are exploited to support tumor immune evasion and growth. By augmenting Tregs’ activities, the PD-1/PD-L1 pathway further suppresses Teffs’ anti-tumor activity, fostering an immune-suppressive milieu that facilitates tumor survival and progression [81]. The potential mechanism of action of PD-1/PD-L1 in cells is shown in Fig. 2.

The potential mechanism of action of PD-1/PD-L1 in cells. In the tumor immune microenvironment (TIME), various cells such as tumor (stromal) cells, certain antigen-presenting cells (APCs), regulatory T cells (Tregs), and M2 macrophages suppress the biological function of cytotoxic T lymphocytes (CTLs) through PD-1 signaling, leading to their exhaustion and apoptosis. Additionally, Tregs can downregulate the expression of CD80/CD86 costimulatory molecules on APCs and upregulate the expression of free PD-L1, exerting a dual inhibitory effect on CTLs. Furthermore, Tregs can secrete inhibitory cytokines and produce granzyme and perforin, causing damage to CTLs or APCs, thereby promoting tumor development. M2 macrophages also secrete growth factors and cytokines that contribute to tumor development and progression. Moreover, immunosuppressive cytokines are released to inhibit CTL metabolism and function
PD-1/PD-L1 in combination with other immune checkpoint inhibitors
The PD-1/PD-L1 pathway is one of several key immune checkpoints, along with molecules like cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), all essential in managing immune responses [82]. CTLA-4 functions by competitively binding to B7 molecules (CD80/CD86) during the initial T cell activation phase, thereby inhibiting CD28-mediated co-stimulatory signals and suppressing T cell activation [83]. Although the CTLA-4 and PD-1 pathways exhibit unique temporal and spatial characteristics, they can work together under certain conditions to amplify immune suppression. TIM-3, mainly found on exhausted T cells, inhibits T cell function by interacting with its ligand galectin-9 [54]. TIM-3 and PD-1 are commonly co-expressed in chronic infections and tumors, collectively dampening antigen-specific T-cell responses [84]. Lymphocyte activation gene-3 (LAG-3) is another crucial immune checkpoint molecule frequently co-expressed with PD-1 on exhausted T cells [85]. By binding to major histocompatibility complex (MHC) class II molecules, LAG-3 inhibits T-cell activation [53, 86]. The interactions of PD-1 with these immune checkpoints provide a rationale for combined immune therapies. For instance, combining PD-1 inhibitors with CTLA-4 inhibitors has demonstrated enhanced anti-tumor effects in certain cancers [87, 88]. This strategy works by simultaneously releasing constraints imposed by multiple inhibitory pathways, thereby bolstering T-cell responses against tumors.
The mechanisms of PD-1/PD-L1regulation in urological tumors
Regulation of PD-1/PD-L1 transcript levels in urologic malignancies
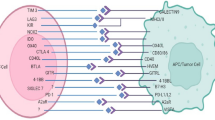
Transcription factors are essential protein molecules that bind specifically to upstream sequences at the 5’ end of genes, thereby regulating the expression of target genes with precise intensity at specific times and in particular spatial locations. Regulation at the transcriptional level plays a pivotal role in determining the level of gene expression. In models pertinent to urogenital tumors, numerous studies have elucidated mechanisms governing mRNA transcription, particularly focusing on PD-1/PD-L1 (especially PD-L1). These findings hold significant value for advancing our understanding of the role and underlying mechanisms of the PD-1/PD-L1 axis in the onset and progression of urogenital malignancies (Fig. 3; Table 1).

Transcriptional regulation of PD-1/PD-L1 in urological malignancies. The key transcriptional mechanisms regulating the PD-1/PD-L1 axis in urological malignancies, including bladder, kidney, and prostate cancers. Highlighted are critical transcription factors (e.g., YY1, HIF-2α, STAT3, NF-κB) and their binding sites on the PD-L1 promoter, which enhance or suppress PD-L1 expression
Several studies have examined the regulatory mechanisms influencing PD-L1 transcriptional levels in prostate cancer (PCa) models. For instance, Yin Yang 1 (YY1), a critical transcriptional regulatory factor, is crucial for tumor cell survival, proliferation, metabolism, epithelial-mesenchymal transition (EMT), and resistance to chemotherapy. YY1 directly interacts with the promoter region of the PD-L1 gene, boosting its transcription. Furthermore, YY1 indirectly influences PD-L1 expression through mechanisms involving p53, cytokines, growth factors, and the PTEN/PI3K/mTOR/AKT pathway. The upregulation of PD-L1 by YY1 significantly decreases the proportion of tumor-infiltrating CD4 + and CD8 + T cells [89]. Another key transcription factor in cancer is NF-κB, which is activated by pro-inflammatory cytokines like TNF-α through the MAP3K7-IKK signaling axis, regulating PD-L1 mRNA expression across various cancers. Huang et al.‘s CHIP-qPCR analysis discovered that the RelA/p65 protein within the NF-κB complex binds to the genomic sites of NF-κB target genes, including PD-L1. Furthermore, electrophoretic mobility shift assay (EMSA) confirmed the presence of a functional NF-κB binding site within the PD-L1 promoter. These findings indicate that NF-κB promotes PD-L1 transcription by directly binding to its promoter, thus facilitating immune evasion in tumors [90].
In BCa models, the transcriptional regulation of PD-L1 has also been extensively studied. c-Jun, part of the AP-1 transcription factor family, can be activated via the classic EGFR/MEK/ERK signaling pathway and binds to the enhancer region of the PD-L1 gene. This activation leads to increased PD-L1 expression and inhibits T-cell functions, identifying c-Jun as a key transcriptional regulator of PD-L1 expression [91]. Additionally, STAT3, a signal transducer and transcription activator, has been associated with the regulation of PD-L1 expression in BCa cells. Lu et al. investigated the interaction between SETD7 and STAT3, finding that STAT3 enhances immune evasion in BCa cells by increasing PD-L1 levels and facilitating tumor immune evasion. These insights suggest that STAT3 directly influences PD-L1 transcription by attaching to its promoter region. Nevertheless, further extensive research is required to confirm these hypotheses [92].
In RCC models, the transcriptional regulation of PD-L1 has been investigated as well. The transcription factor EB (TFEB), known for its role in lysosome biogenesis, autophagy, and metabolism regulation, has been linked to various human tumors. High expression of TFEB is associated with increased tumor cell proliferation and motility. Yang et al. discovered that TFEB directly attaches to the PD-L1 promoter region in RCC, thereby promoting PD-L1 transcription. This process inhibits the cytotoxic activity of tumor-infiltrating CD8 + T cells and enables immune evasion in RCC [93]. Similarly, the transcription factor E3 (TFE3), an essential oncogene in the proliferation of RCC cells, has been demonstrated to enhance PD-L1 expression by binding to its promoter. This regulatory pathway facilitates immune evasion and contributes to sunitinib resistance in RCC [94]. Moreover, hypoxia-inducible factor 2-alpha (HIF2-α), predominantly expressed in RCC cells, plays a significant role in promoting RCC progression. Kim et al. found that HIF2α attaches to hypoxia-response elements on the PD-L1 promoter, resulting in increased PD-L1 transcription. This mechanism also facilitates immune evasion in RCC [95].
Post-transcriptional modification of PD-1/PD-L1 in urologic malignancies
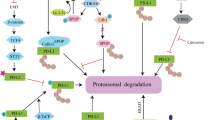
In the context of PD-1/PD-L1, negative regulation occurs through processes like ubiquitination, ubiquitin-like modifications, and methylation (Fig. 4; Table 2). Conversely, positive regulation involves deubiquitination, glycosylation, palmitoylation, ADP-ribosylation, and deacetylation [96]. Understanding these regulatory mechanisms and identifying new targets for modifying PD-1/PD-L1 are crucial for advancing precise immunotherapies for genitourinary malignancies.

Posttranslational modifications of PD-1/PD-L1 in urological malignancies. The various posttranslational modifications (PTMs) of PD-1 and PD-L1 proteins in urological malignancies, including bladder, kidney, and prostate cancers. It highlights modifications such as deubiquitination, methylation, poly-ubiquitination and their specific sites on PD-1/PD-L1 molecules. The figure illustrates how these PTMs affect protein stability, localization, and interaction with other cellular components, ultimately influencing immune checkpoint function
Wang et al. discovered a genetic variant (rs62483508) within the miRNA-responsive element of the long non-coding RNA BCCE4, which is significantly linked to a reduced risk of BCa in the Chinese population. Their research emphasized BCCE4’s role in upregulating USP18 expression by competitively inhibiting miR-328-3p. USP18, a member of the USP subfamily of deubiquitinases, increases PD-L1 protein stability by removing interferon-induced protein 15 (ISG15), which aids in immune escape in BCa cells [97]. Elevated METTL3 expression was also observed in BCa tissues. Further studies indicated that METTL3 expression is regulated through the JNK signaling pathway and the downstream transcription factor c-Jun. METTL3 enhances PD-L1 expression by affecting its methylation status, stabilizing PD-L1 mRNA, and reducing CD8 + T cell cytotoxicity against BCa cells, thereby promoting immune evasion [98]. Additionally, Fibroblast Growth Factor Receptor 3 (FGFR3) has been shown to influence PD-L1 ubiquitination, impacting the tumor microenvironment in BCa. FGFR3 phosphorylates the NEDD4 E3 ubiquitin ligase, facilitating K48-linked polyubiquitination of PD-L1, which leads to its degradation and enhances CD8 + T cell-mediated tumor cell death [99]. Lin et al. investigated the ubiquitination regulatory mechanisms of PD-L1 in BCa, focusing on the E3 ubiquitin ligase RNF144A, which is frequently mutated or epigenetically silenced in cancer and exerts tumor-suppressive effects. Their findings demonstrated that RNF144A interacts with PD-L1 on cell membranes and intracellular vesicles via its C-terminal region, promoting PD-L1 polyubiquitination and degradation. This mechanism enhances CD8 + T cell cytotoxic function and inhibits BCa progression [100].
Li et al. studied the mechanisms behind elevated PD-1 expression in T cells within RCC. They found that transforming growth factor beta 1 (TGFβ1) activates p38, leading to Ser10 phosphorylation of histone H3. This modification recruits RelA/p65, which increases the expression of SRSF3 and SRSF5 in T cells. SRSF3 directly binds to the 3’UTR of PD-1 mRNA through its RNA recognition motifs, enhancing PD-1 mRNA stability and nuclear export, thus increasing PD-1 expression on T cell surfaces. This process inhibits T cell cytotoxicity against RCC cells [101]. Another study by Li et al. highlighted the significant upregulation of miR-224-5p in extracellular vesicles (EVs) derived from RCC patient urine. They found that miR-224-5p suppresses cyclin D1 expression, leading to decreased activity of the cyclin D-CDK4/6 complex. This reduction inhibits the cullin 3-SPOP E3 ubiquitin ligase-mediated pathway responsible for PD-L1 ubiquitination and degradation, resulting in elevated PD-L1 expression levels in RCC cells. RCC cells can transfer these regulatory mechanisms of PD-L1 expression through EVs, enhancing resistance to T cell-mediated cytotoxicity [102].
Molecular mechanisms regulating the expression of PD-1/PD-L1 in urologic cancers
PD-1 and its ligand PD-L1 play a critical role in tumor therapy by effectively regulating anti-tumor immune responses [103]. PD-L1 is found in various tumors, while PD-1 is predominantly located on T cells within tumor tissues [104]. The interaction between PD-L1 and PD-1 forms a molecular barrier that suppresses immune cell cytotoxicity. By modulating the expression of PD-1/PD-L1, it is possible to reactivate immune responses and overcome this immune suppression [105]. Numerous studies have shown the therapeutic benefits of monoclonal antibodies that target PD-1 and PD-L1 in the treatment of urological cancers [106]. Therefore, understanding the regulatory mechanisms of PD-1/PD-L1 expression is essential for optimizing cancer immunotherapy in these malignancies (Fig. 5; Table 3).

The molecular mechanism of regulating PD-1/PD-L1 in urological malignancies. The molecular pathways and mechanisms involved in the regulation of PD-1/PD-L1 expression in urological cancers, including signaling cascades, transcription factors, and posttranslational modifications that contribute to immune evasion and tumor progression
Suppressing the epigenetic regulator EZH2 activates the double-stranded RNA-STING-interferon signaling pathway, resulting in the upregulation of genes related to antigen presentation, Th1 chemokine signals, and interferon responses, including PD-L1. Studies have shown that inhibiting EZH2 directly increases PD-L1 mRNA levels in PCa cell lines and human PCa tissues, thereby enhancing PD-L1 expression and improving the responsiveness of PCa to PD-1 checkpoint inhibitors [107]. In metastatic castration-resistant PCa, researchers found that phosphorylated retinoblastoma protein (pRB) suppresses NF-κB activity by interacting with its RelA/p65 subunit, reducing PD-L1 expression and promoting anti-tumor immune responses. Traditional Chinese medicine CFF-1 has also been reported to inhibit PD-L1 expression in PCa by suppressing the EGFR/JAK1/STAT3 axis, thus impeding cancer progression [108]. Additionally, Mao et al. demonstrated that heterogeneous nuclear ribonucleoprotein L (HnRNP L) enhances YY1 stability by binding to YY1 mRNA, leading to increased YY1 expression. YY1, a transcriptional regulator, binds to the PD-L1 gene promoter, promoting PD-L1 transcription and inhibiting T-cell cytotoxicity [89]. Another study indicated that Neuropeptide-2 activates Rac1, promoting YAP/TAZ nuclear translocation and transcriptional activity, consequently upregulating PD-L1 expression and suppressing anti-tumor immune responses [109].
The JAK (Janus kinase) signaling pathway is essential for various physiological processes and disease development. This pathway includes JAK1, JAK2, JAK3, and TYK2 kinases, which relay external signals from cell membrane receptors like cytokine and immunoglobulin receptors. When these receptors are activated, JAK proteins get phosphorylated and activated, initiating downstream signaling. Activated JAK proteins then phosphorylate and activate STAT proteins, which move to the nucleus to regulate gene transcription, affecting cell proliferation, differentiation, and apoptosis. Thus, the JAK pathway is crucial for immune regulation, cell growth, and cancer development. Abnormal activation of this pathway is linked to inflammatory diseases, autoimmune disorders, and various cancers. Therefore, understanding and controlling the JAK pathway is vital for treating and preventing these conditions. In BCa, tumor-associated macrophages (TAMs), especially M2-type TAMs, are crucial in promoting immune evasion and tumor progression. M2-type TAMs secrete TGF-β, which induces STAT3 phosphorylation and nuclear translocation, and promotes pyruvate kinase M2 (PKM2) dimerization, collectively enhancing PD-L1 expression. TGF-β also triggers the canonical Smad signaling pathway, which activates the JAK2/STAT3 signaling cascade, directly regulating PD-L1 expression and driving BCa progression [110]. Expression of FBW7, an F-box protein within the Skp1-Cul1-F-box protein ubiquitin ligase complex, is reduced in drug-resistant RCC models and patient samples, and its protein levels inversely associated with nuclear factor of activated T cells 1 (NFAT1) in RCC patients. FBW7 directly interacts with NFAT1 and regulates PD-L1 expression by modulating the JAK-STAT, TNF, and HIF pathways [111]. PD-L1 expression is differentially regulated in clear cell RCC and papillary RCC, with PD-L1 expression in clear cell RCC being dependent on intact IFN-γ signaling, activating JAK2/STAT1/IRF1 to promote PD-L1 transcription. Conversely, PD-L1 expression in papillary RCC is independent of IFN-γ signaling, highlighting distinct mechanisms in different RCC subtypes [95, 112]. Heng et al. also linked elevated MTHFD2 expression in BCa cells and tissues with adverse clinical outcomes, showing that MTHFD2 promotes PD-L1 expression by activating the JAK/STAT signaling pathway, highlighting its role in immune regulation within the BCa microenvironment [113].
The ERK (extracellular signal-regulated kinase) signaling pathway is a vital intracellular mechanism essential for cell proliferation, differentiation, survival, and migration. It is part of the RAS-RAF-MEK-ERK cascade, also known as the MAPK (mitogen-activated protein kinase) pathway. This pathway detects external signals via cell surface receptors, such as growth factor and cytokine receptors. Signal transduction starts with receptor activation, leading to RAS protein activation, which then activates RAF kinase. RAF kinase phosphorylates and activates MEK, which subsequently activates ERK. Activated ERK moves into the nucleus to phosphorylate target proteins, including transcription factors, thus regulating gene expression and cell functions. Dysregulation of the ERK pathway is linked to various diseases, particularly cancer, where it promotes uncontrolled cell proliferation and resistance to apoptosis. The ERK pathway also plays roles in cardiovascular, neurological, and immune diseases. Understanding its regulatory mechanisms is crucial for uncovering the molecular basis of these diseases and developing new treatments. The TME is marked by hypoxia and acidity from glycolysis-induced lactate buildup, facilitating tumor cell migration and invasion [114, 115]. Niu et al. found that glucose metabolism influences PD-L1 expression in RCC via the EGFR/ERK/c-Jun pathway. Elevated PD-L1 expression also upregulates PFKFB3 to regulate glycolysis, suggesting potential combined therapeutic targets in RCC [116]. T-LAK cell-originated Protein Kinase (TOPK) regulates PD-L1 expression to promote immune evasion in RCC through two mechanisms. TOPK forms a positive feedback loop with ERK2 and enhances the TGF-β/Smad pathway by directly binding to Smad4, thereby promoting PD-L1 expression [117]. Glutamine is a versatile nutrient crucial for various metabolic pathways within tumor cells. In the TME, glutamine levels are notably low, often reaching undetectable levels compared to normal tissues [118]. Studies highlight that reduced glutamine metabolism in RCC cells correlates with high PD-L1 expression levels. Mechanistically, glutamine deprivation activates the Epidermal Growth Factor Receptor (EGFR), ERK1/2, and c-Jun, resulting in high PD-L1 expression. In turn, high PD-L1 levels contribute to T-cell suppression and facilitate immune evasion of RCC [119].
The PI3K/AKT pathway is an essential intracellular signaling route that regulates cell growth, proliferation, metabolism, and survival. This pathway involves PI3K and AKT, which respond to signals from various receptors like growth factor, insulin, and cytokine receptors. Activation of these receptors leads to PI3K activation at the cell membrane, converting PIP2 to PIP3. PIP3 then activates AKT, which phosphorylates targets like mTOR, GSK-3β, and BAD, controlling cell functions. Abnormal PI3K/AKT activation is linked to diseases such as cancer, where it promotes rapid growth, apoptosis resistance, increased metabolism, and invasiveness. It also plays roles in diabetes, cardiovascular, and neurodegenerative diseases. Understanding this pathway is essential for disease research and developing new treatments. Methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) has been implicated in various malignancies, including BCa. Higher MTHFD2 expression in BCa tissues is associated with poor prognosis, increased tumor immune cell infiltration, and elevated PD-L1 levels. Transcriptome analysis reveals that MTHFD2 expression correlates positively with the activation of the PI3K/AKT signaling pathway, which enhances PD-L1 expression [120]. TFE3, involved in autophagy and lysosome biogenesis, shows elevated expression and activity in various human cancers, including RCC. TFE3 upregulates PD-L1 expression by activating the PI3K/AKT/mTOR pathway, and mTOR inhibitors may further enhance PD-L1 expression by increasing TFE3 activity [121].
In addition, other pathways also play important roles in regulating PD1/PD-L1. The androgen receptor (AR) positively influences PD-L1 expression; miR-200a-3p targets the 3’UTR region of PD-L1 mRNA to suppress its expression. AR upregulates circ_0001005 by modulating the ADAR2 enzyme, and circ_0001005 acts as a competitive inhibitor of miR-200a-3p, thereby reducing miR-200a-3p levels and increasing PD-L1 expression, diminishing NK cell cytotoxicity and promoting immune evasion in BCa [122]. Finally, Nicotinamide phosphoribosyltransferase (NAMPT) has been identified as a negative regulator of PD-L1 expression in BCa. Upregulated NAMPT levels in BCa patients inversely correlate with immune cell infiltration, and it has been shown that NAMPT downregulates PD-L1 expression via an IFN-γ-dependent mechanism, suggesting NAMPT as a potential target for immune checkpoint regulation [123]. Ribonucleotide Reductase Regulatory Subunit M2 (RRM2) is crucial for deoxyribonucleotide synthesis following ribonucleotide reduction and is considered a proto-oncogene due to elevated expression in various cancers. Jin et al. demonstrated that RRM2 upregulates ANXA1 levels, activation, and subsequent PD-L1 expression enhancement, which augments the anti-tumor efficacy of PD-1 blockade therapy [124]. Growth hormone-inducible transmembrane protein (GHITM), a member of the the Bax inhibitor-1 family, is downregulated in RCC and correlates with adverse patient prognosis. Furthermore, Wang et al. demonstrated that GHITM indirectly inhibits PD-L1 expression by suppressing the oncogene Notch1 thereby reducing RCC tumor progression and lung metastasis, suggesting its potential as a therapeutic target [125].
Anti-PD-1 /PD-L1 combination therapy in urinary malignancies
Because of the inherent variability of cancer and the genetic diversity among individuals, single-agent therapies that target the PD-1/PD-L1 pathway have failed to yield satisfactory results [126]. As a result, there is a pressing need for personalized combination therapies designed for individual patients to enhance response rates to PD-1/PD-L1 inhibitors and to overcome resistance to anti-PD-1/PD-L1 treatments. Recent research highlights that integrating anti-PD-1/PD-L1 therapy with other immune checkpoint inhibitors (ICIs) targeted therapies against VEGF/VEGFR, tumor vaccines, oncolytic viruses (OVs), and approaches that can increase the efficacy of PD-1/PD-L1 blockade [127, 128]. Significantly, these strategies have demonstrated initial success in the treatment of genitourinary malignancies (Fig. 6; Table 4).

Combined therapy with anti-PD-1/PD-L1 in the treatment of urological malignancies. Co-administration of anti-PD-1/PD-L1 therapies in urological malignancies demonstrates enhanced antitumor efficacy
LAG-3 is an immune checkpoint inhibitor primarily found on activated T-cells, which curtails T-cell function and proliferation, thereby reducing anti-tumor immune responses. In mouse models of prostate cancer, simultaneous blocking of PD-1 and LAG-3 results in increased infiltration of CD4 + and CD8 + T cells into tumors. Specifically, CD8 + T cells predominantly exhibit effector memory and tissue-resident memory traits. This combination therapy markedly suppresses tumor growth compared to the use of PD-1 or LAG-3 inhibitors alone [129]. Tim-3, co-expressed with PD-1 in TME, represents a marker of T-cell exhaustion and research shows that blocking PD-1 increases Tim-3 levels. Using inhibitors targeting both PD-1 and Tim-3 enhances anti-tumor immunity by improving CD8 + T cell activity, diminishing tumor-supportive factors, and further reducing tumor growth [130]. Nitroxoline, commonly utilized for urinary tract infection treatment, has shown promise as an anticancer agent effective against multiple cancer forms [131, 132]. Huang et al.‘s research demonstrate that Nitroxoline, when combined with PD-1 blockade, shows significant potential in treating PCa. Nitroxoline inhibits the PI3K/AKT/mTOR pathway to reduce PD-L1 expression, thereby boosting the immune response of PD-1 blockade. This therapeutic approach increases memory T cells and decreases myeloid-derived suppressor cells, presenting a promising treatment option [133]. Gene therapy can be used to stimulate immune responses within the TME by directly injecting virus vectors carrying specific genes into tumor sites. For example, the replication-deficient recombinant adenovirus vector rAd-p53, which carries the human p53 gene, boosts the infiltration of CD4 + and CD8 + T cells into tumors and increases PD-L1 expression on tumor cells, thus improving the effectiveness of anti-PD-1 antibody therapy [134]. Docetaxel, an FDA-approved taxane used for prostate cancer (PCa) chemotherapy, has been studied in conjunction with PD-1 inhibitors in PCa mouse models. This combined therapy mitigates the docetaxel-induced increase in PD-L1 expression, reversing tumor immune evasion. Furthermore, it enhances the infiltration of CD4 + and CD8 + T cells into tumors, restores immune functionality, and inhibits the PI3K/AKT/NF-κB-P65/PD-L1 signaling pathway. Consequently, it overcomes chemotherapy resistance induced by docetaxel and promotes apoptosis in tumor cells [135]. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) anchored vaccine represents a novel immunotherapy method. By fixing GM-CSF protein on tumor cell surfaces using a biotin-avidin system, this approach stimulates bone marrow cell proliferation and enhances anti-tumor immune responses. Infiltrating B lymphocytes within the PCa TME exhibit immunosuppressive properties that promote tumor growth. Bruton’s tyrosine kinase (BTK), a critical regulator of B cell function, is targeted by the inhibitor ibrutinib, which demonstrates anti-tumor effects in PCa. While monotherapy with ibrutinib reduces B cell infiltration and activation in the TME without shrinking tumors, combined therapy of ibrutinib and anti-PD-1 ICIs significantly enhances anti-tumor immune responses and reduces tumor volume in preclinical models [136].
T-cell immunoglobulin and ITIM domain (TIGIT) is a negative immune checkpoint receptor found on activated T cells. Its interaction with the ligand CD155 leads to diminished anti-tumor activity of CD8 + T cells by inducing exhaustion. Studies show that blocking TIGIT boosts cytokine production by CD8 + tumor-infiltrating lymphocytes (TILs) and works in tandem with PD-1 blockade in breast cancer (BCa) patients. This indicates that TIGIT and PD-1 jointly inhibit CD8 + T cell effector function, supporting the combined blockade to revive anti-tumor responses [137]. CTLA-4, another inhibitory immune checkpoint receptor present on activated T cells, suppresses their activation and proliferation. Within the BCa TME, CTLA-4 aids in immune evasion. Simultaneously blocking PD-1 and CTLA-4 in mouse BCa models significantly curtails tumor growth by enhancing Th1-type immune responses and triggering IFN-γ signaling pathways in endothelial cells [138]. To combat resistance to PD-1/PD-L1 therapy in bladder cancer, Dumontet and colleagues created a resistant mouse model and investigated combination therapy targets. They found that targeting TNF-α, CD47, and Ly6G in conjunction with anti-PD-1 treatment significantly inhibited tumor growth and overcame resistance more effectively than anti-PD-1 monotherapy [139]. Oxaliplatin (OXP), a second-line chemotherapy for bladder cancer (BCa), when paired with anti-PD-1 inhibitors, demonstrates superior efficacy in reducing tumor growth compared to either treatment used individually. This therapy combination improves immune cell infiltration into the tumor microenvironment (TME) and adjusts immune cell phenotypes to strengthen anti-tumor immune responses [140]. S100A5, a tumor-specific protein, inhibits CD8 + T cell proliferation and cytotoxicity, thereby promoting tumor survival. Indeed, knocking down S100A5 or using anti-PD-1 monotherapy boosts CD8 + T cell infiltration and cytotoxicity, significantly hindering tumor growth and a combination of S100A5 knockdown along with anti-PD-1 therapy demonstrates superior anti-tumor efficacy [141]. Fibroblast Growth Factor Receptor (FGFR) mutations activate Tregs in the TME, impeding anti-PD-1 therapy efficacy. Erdafitinib, an FGFR inhibitor, reverses this immunosuppressive effect. Combining Erdafitinib with PD-1 inhibitors enhances CD8 + T cell infiltration, augments anti-tumor immune response, and therapeutic outcomes [142]. VAX014, an engineered bacterial minicell-based oncolytic agent, selectively targets and kills α3β1 and α5β1 integrin-expressing tumor cells, inducing anti-tumor immune responses. Intravesical VAX014 combined with systemic PD-L1 blockade enhances systemic anti-tumor immunity, prolonging survival by increasing Th1 cells and activating CD8 + T cells among TILs [143].
αKG (alpha-ketoglutarate) serves as a critical intermediate in the tricarboxylic acid (TCA) cycle. In RCC tissues, αKG levels are markedly reduced compared to normal tissues and inversely correlate with tumor malignancy. This suggests a protective role for αKG in inhibiting tumor initiation and progression. Research by Wang et al. further demonstrated that αKG alone or in combination with anti-PD-1 antibodies can inhibit tumor growth, as expected combination therapy was shown to be more effective than monotherapy. By modulating the αKG-B2M-CD8 axis, combination therapy enhances the infiltration of CD8 + T cells into the TME and extends overall survival in murine models [144]. Renetinib, a multi-target tyrosine kinase inhibitor that inhibits VEGFR1-3, FGFR1-4, PDGFR-α, RET, and KIT, has been approved for advanced RCC treatment. Studies show that Renetinib enhances tumor cell sensitivity to IFN-γ signaling by inhibiting the FGFR pathway. Preclinical models demonstrate that using a combination of Renetinib and anti-PD-1 antibodies significantly decreases tumor size and extends survival more effectively than anti-PD-1 monotherapy [145]. Sunitinib, a small molecule inhibitor targeting VEGFRs, demonstrates efficacy in RCC by prolonging survival and inhibiting metastasis. However, its efficacy alone may be limited. Research indicates that combining Sunitinib with anti-PD-L1 therapy is more effective than either treatment alone, significantly extending survival and suppressing lung metastasis [146]. PD-1 blockade leads to increased LAG-3 expression on CD4 + and CD8 + T cells. Combined PD-1 and LAG-3 blockade enhances IFN-γ secretion by CD8 + T cells and improves the overall functionality of TILs compared to PD-1 blockade alone [147]. JX-594, a genetically modified oncolytic virus expressing GM-CSF, activates dendritic cells and induces anti-tumor effects through oncolysis and immune stimulation. Combination therapy with JX-594 and PD-1 inhibitors in metastatic RCC reshapes the tumor immune microenvironment, enhances anti-tumor immune responses, and reduces immune-related toxicity, demonstrating efficacy and safety in preclinical models. Acarbose, an inhibitor of oligosaccharide hydrolysis, modulates glucose metabolism in the TME, enhancing CD8 + T cell anti-tumor immune function. Combination therapy with acarbose and anti-PD-1 significantly enhances the cytotoxic function of CD8 + T cells infiltrating tumors, inhibits RCC tumor growth, and reduces lung metastases compared to anti-PD-1 monotherapy [148].
The clinical trial of PD-1/PD-L1 inhibitors in urological tumors
In the domain of urological cancers such as BCa, RCC, and PCa, PD-1/PD-L1 inhibitors have become essential treatment options. BCa, in particular, has gained significant attention, as illustrated by the IMvigor210 trial, which assessed the efficacy of the PD-L1 inhibitor Atezolizumab in patients with advanced urothelial carcinoma after chemotherapy. The trial revealed notable anti-tumor effects, achieving an objective response rate (ORR) of 15% [149, 150]. Similarly, KEYNOTE-045 investigated Pembrolizumab, a PD-1 inhibitor, showing improved overall survival compared to chemotherapy in advanced BCa [151, 152]. Promising results have been observed with PD-1/PD-L1 inhibitors in advanced RCC treatment. The CheckMate 025 trial assessed Nivolumab in patients with advanced RCC, revealing better overall survival and higher ORRs compared to Everolimus [153, 154]. Moreover, the CheckMate 214 study investigated the combination of Nivolumab and the CTLA-4 inhibitor Ipilimumab, yielding notable outcomes in patients with intermediate and poor risk [155]. PCa exhibits limited response to PD-1/PD-L1 inhibitors overall, though promising efficacy has been observed in specific subgroups. The KEYNOTE-199 trial in metastatic castration-resistant PCa revealed modest overall ORRs in patients treated with Pembrolizumab but efficacy in subgroups carrying DNA repair gene mutations [156]. Future research directions include optimizing PD-1/PD-L1 inhibitor applications through combination therapies, identifying patient subgroups most likely to benefit, and expanding exploration into their potential across urological cancers. While significant strides have been made, further investigations are crucial to fully harnessing their therapeutic potential in clinical practice. The ongoing clinical trials exploring the efficacy of anti-PD-1 and anti-PD-L1 in urological malignancies are shown in Tables 5 and 6.
Discussion and prospects
The use of PD-1 and PD-L1 inhibitors in the treatment of malignant urinary system tumors presents significant challenges due to the development of resistance that needs to be overcome. Central to this issue is the TME, which includes diverse cell types such as TAMs, TILs, and fibroblasts [33, 157]. These components alter immune dynamics by secreting cytokines and growth factors, influencing the effectiveness of PD-1/PD-L1 inhibitors. Resistance also stems from specific genetic mutations and aberrant signaling pathways. For instance, mutations in JAK1/2 genes can disrupt IFN-γ signaling, enabling tumor cells to evade immune surveillance [158]. Furthermore, dysregulated activation of the PI3K/AKT/mTOR pathway is associated with resistance to PD-1/PD-L1 inhibitors. The regulation of PD-L1 expression plays a pivotal role in determining the efficacy of PD-1/PD-L1 inhibitors. Tumor cells can also modulate PD-L1 expression through genomic and epigenomic mechanisms [159]. Epigenetic modifications such as DNA methylation and histone alterations enable tumor cells to reduce PD-L1 gene expression, thereby enhancing resistance to inhibitor therapies. Additionally, tumors employ various strategies to evade immune detection, including elevated expression of immune checkpoint molecules, secretion of immunosuppressive factors, and stimulation of immune-suppressive cell proliferation [52, 160]. Collectively, these mechanisms contribute to resistance against PD-1/PD-L1 inhibitors. Combining PD-1/PD-L1 inhibitors with other ICIs represents a promising strategy to bolster anti-tumor immune responses and surmount resistance encountered with single-agent therapies. By targeting multiple immune regulatory pathways simultaneously, combination therapies hold the potential to enhance treatment outcomes in patients with urinary system malignancies.
Combining PD-1/PD-L1 inhibitors with various treatment modalities enhances anti-tumor effects in urinary system cancers by leveraging complementary mechanisms. Radiotherapy induces tumor cell apoptosis and releases antigens, priming the immune system to recognize tumors, synergizing with PD-1/PD-L1 inhibitors [52, 161]. Emerging therapies like cancer vaccines, OVs, and CAR-T cell therapy boost immune responses through distinct mechanisms, such as direct tumor cell lysis and antigen release [88, 162]. Targeted therapies specific to the TME, such as TAM inhibitors, anti-angiogenics, and TGF-β pathway inhibitors, reduce immunosuppression in the TME, enhancing PD-1/PD-L1 inhibitor efficacy [163]. Gene editing tools like CRISPR/Cas9 address resistance by manipulating genes responsible, restoring immune recognition of tumors [164]. Epigenetic regulators like DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi) alter gene expression, sensitizing tumors to immune attack and reversing immune suppression [50, 165]. These approaches collectively overcome resistance, offering avenues to improve clinical outcomes with PD-1/PD-L1 inhibitors in urinary system cancers.
Personalized medicine offers significant promise in treating urinary system tumors by enhancing treatment efficacy through tailored therapeutic approaches. Genomic sequencing identifies specific mutations, gene expression patterns, and genomic instability in tumors, crucial for predicting responses to PD-1 and PD-L1 ICI. Proteomic analysis examines protein expression and modifications in tumors and their microenvironment, providing insights into tumor biology and potential targets for therapy. Understanding PD-L1 expression and its regulation in the TME optimizes combination immunotherapy strategies. High-throughput screening technologies like single-cell sequencing and mass spectrometry provide detailed insights into tumor heterogeneity and the immune microenvironment, guiding precise treatment planning to improve patient outcomes. Integrating genomic and proteomic data enables personalized treatment regimens tailored to individual molecular profiles, prioritizing PD-1/PD-L1 inhibitors for patients with high PD-L1 expression and considering combined therapies for those with specific mutations or other molecular characteristics.
Liquid biopsy technology, a non-invasive method, analyzes circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) in blood to monitor real-time genomic changes and resistance mechanisms in tumors. This facilitates prompt adjustments to treatment plans, assessment of treatment efficacy, and early detection of recurrence. Personalized therapy requires consideration of the patient’s immune microenvironment, including immune cell infiltration, cytokine expression levels, and immune-suppressive conditions within the TME, all of which significantly impact the effectiveness of PD-1/PD-L1 inhibitors. By comprehensively analyzing these factors, treatment responses can be accurately predicted, optimizing therapeutic strategies. Despite substantial progress in PD-1/PD-L1 inhibitors for treating urinary system cancers, many patients remain unresponsive to current therapies. Hence, attention is increasingly focused on developing next-generation ICIs targeting novel checkpoints. Ongoing research explores new targets such as CTLA-4, TIM-3, LAG-3, and others. CTLA-4 inhibitors, like Ipilimumab, show synergistic effects when combined with PD-1/PD-L1 inhibitors, and was recently approved for advanced RCC treatment. Clinical trials are evaluating the safety and efficacy of inhibitors against other emerging targets such as TIM-3 and LAG-3. Additionally, novel immune therapies like bispecific antibodies and fusion proteins are emerging. These agents simultaneously target multiple immune checkpoints, effectively reversing tumor immune suppression and enhancing treatment outcomes. For example, bispecific antibodies can inhibit both PD-1 and LAG-3, thereby bolstering anti-tumor immune responses. Preclinical studies have demonstrated that LAG-3 inhibitors exhibit significant anti-tumor activity and synergistic effects in combination with PD-1 inhibitors [166,167,168]. Early clinical trials further validate the safety and efficacy of these drugs in patients, supporting their potential therapeutic role. Various combination therapies involving ICIs have also shown promising prospects in early clinical trials. The future of treating urological cancers will likely embrace personalized medicine approaches integrating genomic and proteomic analyses, alongside tailored therapies aligned with individual patient profiles.
The heterogeneity of urological malignancies substantially impacts the effectiveness of PD-L1 and PD-1 blockade therapies [41, 169, 170]. Although these ICIs have demonstrated impressive efficacy in the treatment of BCa and RCC, their utilization in PCa has yet to achieve widespread endorsement. This discrepancy is primarily attributed to the lower immunogenicity of PCa, characterized by a paucity of neoantigens in the TME, thereby impeding the immune system’s ability to identify and eliminate malignant cells (Fig. 7). Conversely, BCa and RCC typically exhibit a higher mutational burden and a more diverse antigenic repertoire, enhancing the responsiveness to ICIs. In BCa and RCC, anti-PD-L1 and PD-1 therapies function by inhibiting the PD-1/PD-L1 axis, thus relieving T cell suppression and augmenting the immune response against neoplasm. However, due to the low immunogenicity and intricate TME of PCa, the response to immunotherapy is often suboptimal. Research indicates that the TME in PCa is infiltrated by a significant number of immunosuppressive cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which secrete inhibitory cytokines to diminish the anti-tumor immune response. Moreover, while certain immunotherapeutic agents have shown substantial efficacy in BCa and RCC, their effectiveness is not uniform across all urological cancers. Analyzing their mechanisms of action suggests that this variability may be linked to the distinct microenvironmental characteristics, mutational landscapes, and immune evasion strategies inherent to different tumor types. Therefore, when evaluating the application of immunotherapy in urological malignancies, it is crucial to consider the heterogeneity of these tumors and tailor therapeutic approaches accordingly. A comprehensive understanding of these differences can facilitate the optimization of treatment protocols, ultimately enhancing patient survival rates and quality of life.

Mutational loads across different tumor types correlate with tumor immunogenicity.Reproduced with permission [171]. Copyright 2018, Springer Nature
Anti-PD-1 and anti-PD-L1 immunotherapies have demonstrated potential in managing urological malignancies, notably BCa and RCC. Nevertheless, their therapeutic efficacy in these cancers is comparatively modest, particularly when juxtaposed with malignancies such as melanoma. PCa exemplifies this disparity, exhibiting lower clinical response rates. The inherent heterogeneity of urological neoplasms and the intricate TME present significant obstacles that attenuate the effectiveness of these therapies. Specifically, the low immunogenicity and immunosuppressive milieu of PCa impede the therapeutic impact of anti-PD-1 and anti-PD-L1 agents, resulting in suboptimal clinical outcomes. To circumvent these challenges, researchers are intensively investigating the potential of combinatory therapeutic strategies. Integrating ICIs with other treatment modalities, including chemotherapy, radiotherapy, targeted therapies, and additional forms of immunotherapy, may potentiate immune responses and augment therapeutic efficacy. For instance, clinical studies have indicated that the concurrent administration of anti-PD-1/PD-L1 agents with platinum-based chemotherapy significantly enhances overall response rates and extends survival in patients with urothelial carcinoma. Moreover, emerging clinical trials are focusing on novel combinatorial approaches to bolster the effectiveness of these therapies. These strategies encompass the integration of immunotherapy with cancer vaccines, adoptive cellular therapies, or other immunomodulatory agents, aiming to robustly activate the patient’s immune system for a more efficacious antitumor response. In summary, although anti-PD-1 and anti-PD-L1 therapies encounter limitations in the treatment of urological malignancies, their therapeutic potential can be further realized through rational combination strategies and innovative clinical trial designs, thereby providing more efficacious treatment avenues for patients.
Conclusion
In this detailed review, we describe the crucial function of the PD-1/PD-L1 pathway in urological malignancies, including BCa, RCC, and PCa. Aberrant PD-1/PD-L1 signaling has been recognized as a significant factor in these cancers, facilitating immune evasion, tumor progression, and metastasis. Both preclinical and clinical studies have repeatedly demonstrated that inhibiting PD-1/PD-L1 interactions boosts anti-tumor immune responses and effectively suppresses tumor growth. These results highlight the therapeutic promise of PD-1/PD-L1 inhibitors in urological cancers.
Data availability
No datasets were generated or analysed during the current study.
Change history
14 September 2024
A Correction to this paper has been published: https://doi.org/10.1186/s12943-024-02121-9
Abbreviations
- PD-1:
-
Programmed death receptor 1
- PD-L1:
-
Programmed death ligand-1
- BCa:
-
Bladder cancer
- RCC:
-
Renal cell carcinoma
- PCa:
-
Prostate cancer
- NMIBC:
-
Non-muscle invasive bladder cancer
- MIBC:
-
Muscle invasive bladder cancer
- ccRCC:
-
Clear cell renal cell carcinoma
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- NK:
-
Natural killer
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- ITSM:
-
immunoreceptor tyrosine-based switch motif
- APCs:
-
Antigen-presenting cells
- TCR:
-
T cell receptor
- NF-κB:
-
Nuclear factor kappa B
- AP-1:
-
Activator protein 1
- IRF:
-
Interferon regulatory factors
- HDACs:
-
Histone deacetylases
- IFN-γ:
-
Including interferon-gamma
- TGF-β:
-
Transforming growth factor-beta
- miRNAs:
-
MicroRNAs
- TME:
-
Tumor microenvironment
- HIF-1α:
-
Hypoxia-inducible factor 1-alpha
- TNF-α:
-
Tumor necrosis factor-alpha
- DCs:
-
Dendritic cells
- DCs:
-
Dendritic cells
- Tregs:
-
Regulatory T cells
- Teffs:
-
Effector T cells
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- TIM-3:
-
T-cell immunoglobulin and mucin domain-containing protein 3
- LAG-3:
-
Lymphocyte activation gene-3
- MHC:
-
Major histocompatibility complex
- YY1:
-
Yin Yang 1
- EMSA:
-
Electrophoretic mobility shift assay
- TFEB:
-
Transcription factor EB
- TFE3:
-
Transcription factor E3
- HIF2α:
-
Hypoxia-inducible factor 2-alpha
- PTMs:
-
Post-transcriptional modifications
- ISG15:
-
Interferon-induced protein 15
- FGFR3:
-
Fibroblast Growth Factor Receptor 3
- TGFβ1:
-
Transforming growth factor beta 1
- EVs:
-
Extracellular vesicles
- pRB:
-
Phosphorylated retinoblastoma protein
- HnRNP L:
-
Heterogeneous nuclear ribonucleoprotein L
- TAMs:
-
Tumor-associated macrophages
- PKM2:
-
Pyruvate kinase M2
- AR:
-
Androgen receptor
- MTHFD2:
-
Methylenetetrahydrofolate dehydrogenase 2
- RRM2:
-
Ribonucleotide Reductase Regulatory Subunit M2
- NFAT1:
-
Nuclear factor of activated T cells 1
- TOPK:
-
T-LAK cell-originated Protein Kinase
- EGFR:
-
Epidermal Growth Factor Receptor
- GHITM:
-
Growth hormone-inducible transmembrane protein
- GM-CSF:
-
Granulocyte-Macrophage Colony-Stimulating Factor
- BTK:
-
Bruton’s tyrosine kinase
- TIGIT:
-
T-cell immunoglobulin and ITIM domain
- BCa:
-
Breast cancer
- OXP:
-
Oxaliplatin
- FGFR:
-
Fibroblast Growth Factor Receptor
- αKG:
-
Alpha-ketoglutarate
- TCA:
-
Tricarboxylic acid
- ORR:
-
Objective response rate
- TILs:
-
Tumor-infiltrating lymphocytes
- Ovs:
-
Oncolytic viruses
- DNMTi:
-
DNA methyltransferase inhibitors
- HDACi:
-
Histone deacetylase inhibitors
- ICI:
-
Immune checkpoint inhibitors
References
Ben-David R, Galsky MD, Sfakianos JP. Novel bladder-sparing approaches in patients with muscle-invasive bladder cancer. Trends Mol Med, (2024).
Guo CC, Lee S, Lee JG, Chen H, Zaleski M, Choi W, McConkey DJ, Wei P, Czerniak B. Molecular profile of bladder cancer progression to clinically aggressive subtypes. Nat Rev Urol, (2024).
Park JS, Lee ME, Kim J, Oh K, Lee N, Jung M, Jang WS, Ham WS. PD-1 inhibitor plus oncolytic vaccinia virus is a safe and effective treatment option for metastatic renal cell carcinoma. Cancer Cell Int. 2024;24:50.
Grobet-Jeandin E, Lenfant L, Pinar U, Parra J, Mozer P, Renard-Penna R, Thibault C, Roupret M, Seisen T. Management of patients with muscle-invasive bladder cancer with clinical evidence of pelvic lymph node metastases. Nat Rev Urol. 2024;21:339–56.
Alfred Witjes J, Max Bruins H, Carrion A, Cathomas R, Comperat E, Efstathiou JA, Fietkau R, Gakis G, Lorch A, Martini A, Mertens LS, Meijer RP, Milowsky MI, Neuzillet Y, Panebianco V, Redlef J, Rink M, Rouanne M, Thalmann GN, Saebjornsen S, Veskimae E, van der Heijden AG. European Association of Urology Guidelines on muscle-invasive and metastatic bladder Cancer: Summary of the 2023 guidelines. Eur Urol. 2024;85:17–31.
Maas M, Todenhofer T, Black PC. Urine biomarkers in bladder cancer - current status and future perspectives. Nat Rev Urol. 2023;20:597–614.
Jubber I, Ong S, Bukavina L, Black PC, Comperat E, Kamat AM, Kiemeney L, Lawrentschuk N, Lerner SP, Meeks JJ, Moch H, Necchi A, Panebianco V, Sridhar SS, Znaor A, Catto JWF, Cumberbatch MG. Epidemiology of bladder Cancer in 2023: a systematic review of risk factors. Eur Urol. 2023;84:176–90.
Dyrskjot L, Hansel DE, Efstathiou JA, Knowles MA, Galsky MD, Teoh J, Theodorescu D. Bladder cancer. Nat Rev Dis Primers. 2023;9:58.
Lopez-Beltran A, Cookson MS, Guercio BJ, Cheng L. Advances in diagnosis and treatment of bladder cancer. BMJ. 2024;384:e076743.
Matuszczak M, Salagierski M. Diagnostic and prognostic potential of biomarkers CYFRA 21.1, ERCC1, p53, FGFR3 and TATI in bladder cancers. Int J Mol Sci, 21 (2020).
Matuszczak M, Schalken JA, Salagierski M. Prostate Cancer Liquid Biopsy Biomarkers’ Clinical Utility in Diagnosis and Prognosis. Cancers (Basel), 13 (2021).
Matuszczak M, Kiljanczyk A, Salagierski M. A liquid biopsy in bladder Cancer-The Current Landscape in urinary biomarkers. Int J Mol Sci, 23 (2022).
Shkolyar E, Zhou SR, Carlson CJ, Chang S, Laurie MA, Xing L, Bowden AK, Liao JC. Optimizing cystoscopy and TURBT: enhanced imaging and artificial intelligence. Nat Rev Urol, (2024).
Matuszczak M, Kiljanczyk A, Salagierski M. The Role of Focal Therapy and Active Surveillance for Small Renal Mass Therapy, Biomedicines, 10 (2022).
Chen J, Huang CP, Quan C, Zu X, Ou Z, Tsai YC, Messing E, Yeh S, Chang C. The androgen receptor in bladder cancer. Nat Rev Urol. 2023;20:560–74.
Cotta BH, Choueiri TK, Cieslik M, Ghatalia P, Mehra R, Morgan TM, Palapattu GS, Shuch B, Vaishampayan U, Van Allen E, Ari Hakimi A, Salami SS. Current Landscape of genomic biomarkers in Clear Cell Renal Cell Carcinoma. Eur Urol. 2023;84:166–75.
Mao X, Wang G, Wang Z, Duan C, Wu X, Xu H. Theranostic lipid nanoparticles for renal cell carcinoma. Adv Mater, (2023) e2306246.
Motzer RJ, Rane PP, Saretsky TL, Pawar D, Martin Nguyen A, Sundaram M, Burgents J, Pandey R, Rudell K. Patient-reported Outcome Measurement and reporting for patients with Advanced Renal Cell Carcinoma: a systematic literature review. Eur Urol. 2023;84:406–17.
Catalano M, Procopio G, Sepe P, Santoni M, Sessa F, Villari D, Nesi G, Roviello G. Tyrosine kinase and immune checkpoints inhibitors in favorable risk metastatic renal cell carcinoma: trick or treat? Pharmacol Ther. 2023;249:108499.
Wang Y, Suarez ER, Kastrunes G, de Campos NSP, Abbas R, Pivetta RS, Murugan N, Chalbatani GM, D’Andrea V, Marasco WA. Evolution of cell therapy for renal cell carcinoma. Mol Cancer. 2024;23:8.
Venkatesh N, Martini A, McQuade JL, Msaouel P, Hahn AW. Obesity and renal cell carcinoma: Biological mechanisms and perspectives. Semin Cancer Biol. 2023;94:21–33.
Weiner AB, Kakani P, Armstrong AJ, Bossi A, Cornford P, Feng F, Kanabur P, Karnes RJ, McKay RR, Morgan TM, Schaeffer EM, Shore N, Tree AC, Spratt DE. Risk stratification of patients with recurrence after primary treatment for prostate Cancer. A Systematic Review, Eur Urol; 2024.
Jiao J, Zhang J, Wen W, Qin W, Chen X. Prostate-specific membrane antigen-targeted surgery in prostate cancer: accurate identification, real-time diagnosis, and precise resection. Theranostics. 2024;14:2736–56.
Hussain M, Fizazi K, Shore ND, Heidegger I, Smith MR, Tombal B, Saad F. Metastatic hormone-sensitive prostate Cancer and combination treatment outcomes: a review. JAMA Oncol. 2024;10:807–20.
Cao PHA, Dominic A, Lujan FE, Senthilkumar S, Bhattacharya PK, Frigo DE, Subramani E. Unlocking ferroptosis in prostate cancer - the road to novel therapies and imaging markers. Nat Rev Urol, (2024).
Zhu L, Pan J, Mou W, Deng L, Zhu Y, Wang Y, Pareek G, Hyams E, Carneiro BA, Hadfield MJ, El-Deiry WS, Yang T, Tan T, Tong T, Ta N, Zhu Y, Gao Y, Lai Y, Cheng L, Chen R, Xue W. Harnessing artificial intelligence for prostate cancer management. Cell Rep Med. 2024;5:101506.
Chen H, Pang B, Zhou C, Han M, Gong J, Li Y, Jiang J. Prostate cancer-derived small extracellular vesicle proteins: the hope in diagnosis, prognosis, and therapeutics. J Nanobiotechnol. 2023;21:480.
Gebrael G, Fortuna GG, Sayegh N, Swami U, Agarwal N. Advances in the treatment of metastatic prostate cancer. Trends Cancer. 2023;9:840–54.
Lin X, Kang K, Chen P, Zeng Z, Li G, Xiong W, Yi M, Xiang B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer. 2024;23:108.
Li Y, Sharma A, Schmidt-Wolf IGH. Evolving insights into the improvement of adoptive T-cell immunotherapy through PD-1/PD-L1 blockade in the clinical spectrum of lung cancer. Mol Cancer. 2024;23:80.
Groeger S, Meyle J. The role of programmed death receptor (PD-)1/PD-ligand (L)1 in periodontitis and cancer, Periodontol 2000, (2024).
Hashimoto M, Ramalingam SS, Ahmed R. Harnessing CD8 T cell responses using PD-1-IL-2 combination therapy. Trends Cancer. 2024;10:332–46.
Zhang H, Liu L, Liu J, Dang P, Hu S, Yuan W, Sun Z, Liu Y, Wang C. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. 2023;22:58.
Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, Sun Z, Zhang Y, Wang C. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. 2023;22:29.
Sordo-Bahamonde C, Lorenzo-Herrero S, Granda-Díaz R, Martínez-Pérez A, Aguilar-García C, Rodrigo JP, García-Pedrero JM, Gonzalez S. Beyond the anti-PD-1/PD-L1 era: promising role of the BTLA/HVEM axis as a future target for cancer immunotherapy. Mol Cancer. 2023;22:142.
Liu S, Wang H, Shao X, Chen H, Chao S, Zhang Y, Gao Z, Yao Q, Zhang P. Advances in PD-1 signaling inhibition-based nano-delivery systems for tumor therapy. J Nanobiotechnol. 2023;21:207.
Pang K, Shi ZD, Wei LY, Dong Y, Ma YY, Wang W, Wang GY, Cao MY, Dong JJ, Chen YA, Zhang P, Hao L, Xu H, Pan D, Chen ZS, Han CH. Research progress of therapeutic effects and drug resistance of immunotherapy based on PD-1/PD-L1 blockade. Drug Resist Updat. 2023;66:100907.
Laba S, Mallett G, Amarnath S. The depths of PD-1 function within the tumor microenvironment beyond CD8(+) T cells. Semin Cancer Biol. 2022;86:1045–55.
Chu X, Tian W, Wang Z, Zhang J, Zhou R. Co-inhibition of TIGIT and PD-1/PD-L1 in Cancer Immunotherapy: mechanisms and clinical trials. Mol Cancer. 2023;22:93.
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, Xia Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15:24.
Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28.
Flaig TW, Spiess PE, Abern M, Agarwal N, Bangs R, Boorjian SA, Buyyounouski MK, Chan K, Chang S, Friedlander T, Greenberg RE, Guru KA, Herr HW, Hoffman-Censits J, Kishan A, Kundu S, Lele SM, Mamtani R, Margulis V, Mian OY, Michalski J, Montgomery JS, Nandagopal L, Pagliaro LC, Parikh M, Patterson A, Plimack ER, Pohar KS, Preston MA, Richards K, Sexton WJ, Siefker-Radtke AO, Tollefson M, Tward J, Wright JL, Dwyer MA, Cassara CJ, Gurski LA. NCCN Guidelines(R) Insights: Bladder Cancer, Version 2.2022, J Natl Compr Canc Netw, 20 (2022) 866–878.
Sperger JM, Emamekhoo H, McKay RR, Stahlfeld CN, Singh A, Chen XE, Kwak L, Gilsdorf CS, Wolfe SK, Wei XX, Silver R, Zhang Z, Morris MJ, Bubley G, Feng FY, Scher HI, Rathkopf D, Dehm SM, Choueiri TK, Halabi S, Armstrong AJ, Wyatt AW, Taplin ME, Zhao SG, Lang JM. Prospective evaluation of clinical outcomes using a Multiplex Liquid Biopsy Targeting Diverse Resistance mechanisms in metastatic prostate Cancer. J Clin Oncol. 2021;39:2926–37.
Matuszczak M, Kiljanczyk A, Salagierski M. Surgical Approach in metastatic renal cell carcinoma: a Literature Review. Cancers (Basel), 15 (2023).
Marvaso G, Jereczek-Fossa BA, Zaffaroni M, Vincini MG, Corrao G, Andratschke N, Balagamwala EH, Bedke J, Blanck O, Capitanio U, Correa RJM, De Meerleer G, Franzese C, Gaeta A, Gandini S, Garibaldi C, Gerszten PC, Gillessen S, Grubb WR, Guckenberger M, Hannan R, Jhaveri PM, Josipovic M, Kerkmeijer LGW, Lehrer EJ, Lindskog M, Louie AV, Nguyen QN, Ost P, Palma DA, Procopio G, Rossi M, Staehler M, Tree AC, Tsang YM, Van As N, Zaorsky NG, Zilli T, Pasquier D, Siva S. Delphi consensus on stereotactic ablative radiotherapy for oligometastatic and oligoprogressive renal cell carcinoma-a European Society for Radiotherapy and Oncology study endorsed by the European Association of Urology, Lancet Oncol, 25 (2024) e193-e204.
Bamias A, Davis ID, Galsky MD, Arranz JA, Kikuchi E, Grande E, Del Muro XG, Park SH, De Giorgi U, Alekseev B, Mencinger M, Izumi K, Schutz FA, Puente J, Li JR, Panni S, Gumus M, Ozguroglu M, Mariathasan S, Poloz Y, Bene-Tchaleu F, Lee C, Bernhard S, De Santis M. Atezolizumab monotherapy versus chemotherapy in untreated locally advanced or metastatic urothelial carcinoma (IMvigor130): final overall survival analysis from a randomised, controlled, phase 3 study. Lancet Oncol. 2024;25:46–61.
Powles T, Bellmunt J, Comperat E, De Santis M, Huddart R, Loriot Y, Necchi A, Valderrama BP, Ravaud A, Shariat SF, Szabados B, van der Heijden MS, Gillessen S. E.G.C.E.a. clinicalguidelines@esmo.org, bladder cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33:244–58.
Islam MK, Stanslas J. Peptide-based and small molecule PD-1 and PD-L1 pharmacological modulators in the treatment of cancer. Pharmacol Ther. 2021;227:107870.
Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv, 6 (2020).
Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, Yang B, Ding L. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–33.
Moser JC, Hu-Lieskovan S. Mechanisms of resistance to PD-1 checkpoint blockade. Drugs. 2020;80:459–65.
Yamaguchi H, Hsu JM, Sun L, Wang SC, Hung MC. Advances and prospects of biomarkers for immune checkpoint inhibitors. Cell Rep Med, (2024) 101621.
Sun Q, Hong Z, Zhang C, Wang L, Han Z, Ma D. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct Target Ther. 2023;8:320.
Cai L, Li Y, Tan J, Xu L, Li Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. 2023;16:101.
Pulanco MC, Madsen AT, Tanwar A, Corrigan DT, Zang X. Recent advancements in the B7/CD28 immune checkpoint families: new biology and clinical therapeutic strategies. Cell Mol Immunol. 2023;20:694–713.
Lin Q, Wang X, Hu Y. The opportunities and challenges in immunotherapy: insights from the regulation of PD-L1 in cancer cells. Cancer Lett. 2023;569:216318.
Kiriyama Y, Nochi H. Regulation of PD-L1 expression by Nuclear receptors. Int J Mol Sci, 24 (2023).
Beenen AC, Sauerer T, Schaft N, Dorrie J. Beyond Cancer: regulation and function of PD-L1 in Health and Immune-Related diseases. Int J Mol Sci, 23 (2022).
Liu Z, Yu X, Xu L, Li Y, Zeng C. Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol. 2022;11:44.
Nihira NT, Miki Y. Regulation of intrinsic functions of PD-L1 by post-translational modification in tumors. Front Oncol. 2022;12:825284.
Qu L, Jin J, Lou J, Qian C, Lin J, Xu A, Liu B, Zhang M, Tao H, Yu W. The nuclear transportation of PD-L1 and the function in tumor immunity and progression. Cancer Immunol Immunother. 2022;71:2313–23.
Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol. 2022;19:287–305.
Hu X, Lin Z, Wang Z, Zhou Q. Emerging role of PD-L1 modification in cancer immunotherapy. Am J Cancer Res. 2021;11:3832–40.
Schafer VS, Brossart P, Warrington KJ, Kurts C, Sendtner GW, Aden CA. The role of autoimmunity and autoinflammation in giant cell arteritis: a systematic literature review. Autoimmun Rev. 2023;22:103328.
Ashrafizadeh M, Zarrabi A, Hushmandi K, Zarrin V, Moghadam ER, Zabolian A, Tavakol S, Samarghandian S, Najafi M. PD-1/PD-L1 axis regulation in cancer therapy: the role of long non-coding RNAs and microRNAs. Life Sci. 2020;256:117899.
Liu J, Wu M, Yang Y, Wang Z, He S, Tian X, Wang H. Gammadelta T cells and the PD-1/PD-L1 axis: a love-hate relationship in the tumor microenvironment. J Transl Med. 2024;22:553.
Khosravi GR, Mostafavi S, Bastan S, Ebrahimi N, Gharibvand RS, Eskandari N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun (Lond). 2024;44:521–53.
Zhang J, Fan J, Zeng X, Nie M, Luan J, Wang Y, Ju D, Yin K. Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharm Sin B. 2021;11:609–20.
Ji Z, Shen J, Lan Y, Yi Q, Liu H. Targeting signaling pathways in osteosarcoma: mechanisms and clinical studies. MedComm. 2020;4(2023):e308.
Li C, Cai C, Xu D, Chen X, Song J. Activation, signaling, cancer and therapy. Pharmacol Res. 2024;TREM1:107212.
He R, Huang S, Lu J, Su L, Gao X, Chi H. Unveiling the immune symphony: decoding colorectal cancer metastasis through immune interactions. Front Immunol. 2024;15:1362709.
Yao P, Liang S, Liu Z, Xu C. A review of natural products targeting tumor immune microenvironments for the treatment of lung cancer. Front Immunol. 2024;15:1343316.
He Y, Zhu M, Lai X, Zhang H, Jiang W. The roles of PD-L1 in the various stages of tumor metastasis. Cancer Metastasis Rev, (2024).
Lu MM, Yang Y. Exosomal PD-L1 in cancer and other fields: recent advances and perspectives. Front Immunol. 2024;15:1395332.
Fan Z, Wu C, Chen M, Jiang Y, Wu Y, Mao R, Fan Y. The generation of PD-L1 and PD-L2 in cancer cells: from nuclear chromatin reorganization to extracellular presentation. Acta Pharm Sin B. 2022;12:1041–53.
Bailly C. Regulation of PD-L1 expression on cancer cells with ROS-modulating drugs. Life Sci. 2020;246:117403.
Yang M, Cui M, Sun Y, Liu S, Jiang W. Mechanisms, combination therapy, and biomarkers in cancer immunotherapy resistance. Cell Commun Signal. 2024;22:338.
Wang Z, Zhang J, An F, Zhang J, Meng X, Liu S, Xia R, Wang G, Yan C. The mechanism of dendritic cell-T cell crosstalk in rheumatoid arthritis. Arthritis Res Ther. 2023;25:193.
Vaseq R, Sharma A, Li Y, Schmidt-Wolf IGH. Revising the Landscape of Cytokine-Induced Killer Cell Therapy in Lung Cancer: Focus on Immune Checkpoint inhibitors. Int J Mol Sci, 24 (2023).
Tang W, Chen J, Ji T, Cong X. TIGIT, a novel immune checkpoint therapy for melanoma. Cell Death Dis. 2023;14:466.
Dong S, Guo X, Han F, He Z, Wang Y. Emerging role of natural products in cancer immunotherapy. Acta Pharm Sin B. 2022;12:1163–85.
Qiu J, Cheng Z, Jiang Z, Gan L, Zhang Z, Xie Z. Immunomodulatory Precision: a narrative review exploring the critical role of Immune checkpoint inhibitors in Cancer Treatment. Int J Mol Sci, 25 (2024).
Adeniran AJ, Shuch B, Humphrey PA. Sarcomatoid and Rhabdoid Renal Cell Carcinoma: clinical, pathologic, and Molecular Genetic features. Am J Surg Pathol. 2024;48:e65–88.
Merhi M, Ahmad F, Taib N, Inchakalody V, Uddin S, Shablak A, Dermime S. The complex network of transcription factors, immune checkpoint inhibitors and stemness features in colorectal cancer: a recent update. Semin Cancer Biol. 2023;89:1–17.
Kreidieh FY, Tawbi HA. The introduction of LAG-3 checkpoint blockade in melanoma: immunotherapy landscape beyond PD-1 and CTLA-4 inhibition. Ther Adv Med Oncol. 2023;15:17588359231186027.
Chavanton A, Mialhe F, Abrey J, Baeza Garcia A, Garrido C. LAG-3: recent developments in combinational therapies in cancer. Cancer Sci, (2024).
Zabeti Touchaei A, Vahidi S. MicroRNAs as regulators of immune checkpoints in cancer immunotherapy: targeting PD-1/PD-L1 and CTLA-4 pathways. Cancer Cell Int. 2024;24:102.
Burke KP, Chaudhri A, Freeman GJ, Sharpe AH. The B7:CD28 family and friends: unraveling coinhibitory interactions. Immunity. 2024;57:223–44.
Zhou X, Zou L, Liao H, Luo J, Yang T, Wu J, Chen W, Wu K, Cen S, Lv D, Shu F, Yang Y, Li C, Li B, Mao X. Abrogation of HnRNP L enhances anti-PD-1 therapy efficacy via diminishing PD-L1 and promoting CD8(+) T cell-mediated ferroptosis in castration-resistant prostate cancer. Acta Pharm Sin B. 2022;12:692–707.
Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, Ye Z, Ma T, Wu Q, Rodrigues DN, Kohli M, Jimenez R, Wang L, Goodrich DW, de Bono J, Dong H, Wu H, Zhu R, Huang H. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression, Mol Cell, 73 (2019) 22–35 e26.
Wang L, Xu T, Yang X, Liang Z, Zhang J, Li D, Chen Y, Ma G, Wang Y, Liang Y, Niu H. Immunosuppression Induced by glutamine deprivation occurs via activating PD-L1 transcription in bladder Cancer. Front Mol Biosci. 2021;8:687305.
Lv J, Wu Q, Li K, Bai K, Yu H, Zhuang J, Sun H, Yang H, Yang X, Lu Q. Lysine N-methyltransferase SETD7 promotes bladder cancer progression and immune escape via STAT3/PD-L1 cascade. Int J Biol Sci. 2023;19:3744–61.
Zhang C, Duan Y, Xia M, Dong Y, Chen Y, Zheng L, Chai S, Zhang Q, Wei Z, Liu N, Wang J, Sun C, Tang Z, Cheng X, Wu J, Wang G, Zheng F, Laurence A, Li B, Yang XP. TFEB mediates Immune Evasion and Resistance to mTOR inhibition of renal cell carcinoma via induction of PD-L1. Clin Cancer Res. 2019;25:6827–38.
Guo X, Li R, Bai Q, Jiang S, Wang H. TFE3-PD-L1 axis is pivotal for sunitinib resistance in clear cell renal cell carcinoma. J Cell Mol Med. 2020;24:14441–52.
Kong SK, Kim BS, Lim H, Kim HJ, Kim YS. Dissection of PD-L1 promoter reveals differential transcriptional regulation of PD-L1 in VHL mutant clear cell renal cell carcinoma. Lab Invest. 2022;102:352–62.
Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in Cancer Therapy. Cancer Res. 2018;78:6349–53.
Zheng R, Gao F, Mao Z, Xiao Y, Yuan L, Huang Z, Lv Q, Qin C, Du M, Zhang Z, Wang M. LncRNA BCCE4 genetically enhances the PD-L1/PD-1 Interaction in Smoking-related bladder Cancer by modulating mir-328-3p-USP18 signaling. Adv Sci (Weinh). 2023;10:e2303473.
Ni Z, Sun P, Zheng J, Wu M, Yang C, Cheng M, Yin M, Cui C, Wang G, Yuan L, Gao Q, Li Y. JNK Signaling promotes bladder Cancer Immune escape by regulating METTL3-Mediated m6A modification of PD-L1 mRNA. Cancer Res. 2022;82:1789–802.
Jing W, Wang G, Cui Z, Xiong G, Jiang X, Li Y, Li W, Han B, Chen S, Shi B. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-mediated bladder Cancer Immune Surveillance. Cancer Res. 2022;82:114–29.
Ho SR, Lee YC, Ittmann MM, Lin FT, Chan KS, Lin WC. RNF144A deficiency promotes PD-L1 protein stabilization and carcinogen-induced bladder tumorigenesis. Cancer Lett. 2021;520:344–60.
Wu P, Geng B, Chen Q, Zhao E, Liu J, Sun C, Zha C, Shao Y, You B, Zhang W, Li L, Meng X, Cai J, Li X. Tumor Cell-Derived TGFbeta1 attenuates Antitumor Immune activity of T cells via regulation of PD-1 mRNA. Cancer Immunol Res. 2020;8:1470–84.
Qin Z, Hu H, Sun W, Chen L, Jin S, Xu Q, Liu Y, Yu L, Zeng S. Mir-224-5p contained in urinary extracellular vesicles regulates PD-L1 expression by inhibiting cyclin D1 in renal cell carcinoma cells. Cancers (Basel), 13 (2021).
Weber EW, Maus MV, Mackall CL. Emerg Landsc Immune Cell Ther Cell. 2020;181:46–62.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Feng C, Zhang L, Chang X, Qin D, Zhang T. Regulation of post-translational modification of PD-L1 and advances in tumor immunotherapy. Front Immunol. 2023;14:1230135.
Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. Review of Immunotherapy in Urologic Cancer: evolving roles for targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol. 2015;68:267–79.
Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, Qiu X, Canadas I, Roehle K, Heckler M, Calagua C, Ye H, Pantelidou C, Galbo P, Panja S, Mitrofanova A, Wilkinson S, Whitlock NC, Trostel SY, Hamid AA, Kibel AS, Barbie DA, Choudhury AD, Pomerantz MM, Sweeney CJ, Long HW, Einstein DJ, Shapiro GI, Dougan SK, Sowalsky AG, He HH, Freedman ML, Balk SP, Loda M, Labbe DP, Olson BM, Ellis L. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2:444–56.
Zhang Y, Wei Y, Jiang S, Dang Y, Yang Y, Zuo W, Zhu Q, Liu P, Gao Y, Lu S. Traditional Chinese medicine CFF-1 exerts a potent anti-tumor immunity to hinder tumor growth and metastasis in prostate cancer through EGFR/JAK1/STAT3 pathway to inhibit PD-1/PD-L1 checkpoint signaling. Phytomedicine. 2022;99:153939.
Wang M, Wisniewski CA, Xiong C, Chhoy P, Goel HL, Kumar A, Zhu LJ, Li R, St Louis PA, Ferreira LM, Pakula H, Xu Z, Loda M, Jiang Z, Brehm MA, Mercurio AM. Therapeutic blocking of VEGF binding to neuropilin-2 diminishes PD-L1 expression to activate antitumor immunity in prostate cancer. Sci Transl Med. 2023;15:eade5855.
Yu Y, Liang Y, Xie F, Zhang Z, Zhang P, Zhao X, Zhang Z, Liang Z, Li D, Wang L, Chen Y, Sun L, Niu H, Wang Y. Tumor-associated macrophage enhances PD-L1-mediated immune escape of bladder cancer through PKM2 dimer-STAT3 complex nuclear translocation. Cancer Lett. 2024;593:216964.
Liu W, Ren D, Xiong W, Jin X, Zhu L. A novel FBW7/NFAT1 axis regulates cancer immunity in sunitinib-resistant renal cancer by inducing PD-L1 expression. J Exp Clin Cancer Res. 2022;41:38.
Hanze J, Wegner M, Noessner E, Hofmann R, Hegele A. Co-regulation of Immune Checkpoint PD-L1 with Interferon-Gamma Signaling Is Associated with a Survival Benefit in Renal Cell Cancer. Target Oncol. 2020;15:377–90.
Li L, Zhang Y, Hu W, Zou F, Ning J, Rao T, Ruan Y, Yu W, Cheng F. MTHFD2 promotes PD-L1 expression via activation of the JAK/STAT signalling pathway in bladder cancer. J Cell Mol Med. 2023;27:2922–36.
Kim Y, Lin Q, Glazer PM, Yun Z. Hypoxic tumor microenvironment and cancer cell differentiation. Curr Mol Med. 2009;9:425–34.
Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921–5.
Yu Y, Liang Y, Li D, Wang L, Liang Z, Chen Y, Ma G, Wu H, Jiao W, Niu H. Glucose metabolism involved in PD-L1-mediated immune escape in the malignant kidney tumour microenvironment. Cell Death Discov. 2021;7:15.
Li J, Sun H, Fu M, Zheng Z, Xu C, Yang K, Liu Y, Xuan Z, Bai Y, Zheng J, Zhao Y, Shi Z, Shao C. TOPK mediates immune evasion of renal cell carcinoma via upregulating the expression of PD-L1, iScience, 26 (2023) 107185.
Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol Cell. 2013;50:200–11.
Ma G, Liang Y, Chen Y, Wang L, Li D, Liang Z, Wang X, Tian D, Yang X, Niu H. Glutamine deprivation induces PD-L1 expression via activation of EGFR/ERK/c-Jun Signaling in Renal Cancer. Mol Cancer Res. 2020;18:324–39.
Deng X, Liu X, Hu B, Liu J, Fu B, Zhang W. Upregulation of MTHFD2 is associated with PD–L1 activation in bladder cancer via the PI3K/AKT pathway. Int J Mol Med, 51 (2023).
Lee HJ, Shin DH, Song JS, Park JY, Kim SY, Hwang CS, Na JY, Lee JH, Kim JY, Park SW, Sol MY. mTOR inhibition increases transcription factor E3 (TFE3) activity and modulates programmed death-ligand 1 (PD-L1) expression in translocation renal cell carcinoma. Am J Pathol. 2021;191:1999–2008.
Liu Q, You B, Meng J, Huang CP, Dong G, Wang R, Chou F, Gao S, Chang C, Yeh S, Xu W. Targeting the androgen receptor to enhance NK cell killing efficacy in bladder cancer by modulating ADAR2/circ_0001005/PD-L1 signaling. Cancer Gene Ther. 2022;29:1988–2000.
Chen KC, Dhar T, Chen CR, Chen EC, Peng CC. Nicotinamide phosphoribosyltransferase modulates PD-L1 in bladder cancer and enhances immunotherapeutic sensitivity. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167106.
Xiong W, Zhang B, Yu H, Zhu L, Yi L, Jin X. RRM2 regulates sensitivity to Sunitinib and PD-1 blockade in Renal Cancer by stabilizing ANXA1 and activating the AKT pathway. Adv Sci (Weinh). 2021;8:e2100881.
Huang S, Liu J, Hu J, Hou Y, Hu M, Zhang B, Luo H, Fu S, Chen Y, Liu X, Chen Z, Wang L. GHITM regulates malignant phenotype and sensitivity to PD-1 blockade of renal cancer cells via notch signalling. J Cell Mol Med. 2024;28:e18290.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61.
Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, Schuebbe G, Renz BW, D’Haese JG, Schloesser H, Heinemann V, Subklewe M, Boeck S, von Werner J. Bergwelt-Baildon, advances in cancer immunotherapy 2019 - latest trends. J Exp Clin Cancer Res. 2019;38:268.
Jiang Y, Chen M, Nie H, Yuan Y. PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccin Immunother. 2019;15:1111–22.
Zahm CD, Moseman JE, Delmastro LE, D GM. PD-1 and LAG-3 blockade improve anti-tumor vaccine efficacy. Oncoimmunology. 2021;10:1912892.
Zhang X, Chen H, Li G, Zhou X, Shi Y, Zou F, Chen Y, Gao J, Yang S, Wu S, Long Z. Increased Tim-3 expression on TILs during treatment with the anchored GM-CSF vaccine and anti-PD-1 antibodies is inversely correlated with response in prostate cancer. J Cancer. 2020;11:648–56.
Chang WL, Hsu LC, Leu WJ, Chen CS, Guh JH. Repurposing of Nitroxoline as a potential anticancer agent against human prostate cancer: a crucial role on AMPK/mTOR signaling pathway and the interplay with Chk2 activation. Oncotarget. 2015;6:39806–20.
Mao H, Du Y, Zhang Z, Cao B, Zhao J, Zhou H, Mao X. Nitroxoline shows antimyeloma activity by targeting the TRIM25/p53 axle. Anticancer Drugs. 2017;28:376–83.
Xu N, Huang L, Li X, Watanabe M, Li C, Xu A, Liu C, Li Q, Araki M, Wada K, Nasu Y, Huang P. The Novel combination of Nitroxoline and PD-1 blockade, exerts a potent Antitumor Effect in a mouse model of prostate Cancer. Int J Biol Sci. 2019;15:919–28.
Kunimura N, Kitagawa K, Sako R, Narikiyo K, Tominaga S, Bautista DS, Xu W, Fujisawa M, Shirakawa T. Combination of rAd-p53 in situ gene therapy and anti-PD-1 antibody immunotherapy induced anti-tumor activity in mouse syngeneic urogenital cancer models. Sci Rep. 2020;10:17464.
Zhou S, Wang B, Wei Y, Dai P, Chen Y, Xiao Y, Xia H, Chen C, Yin W. PD-1 inhibitor combined with Docetaxel exerts synergistic anti-prostate cancer effect in mice by down-regulating the expression of PI3K/AKT/NFKB-P65/PD-L1 signaling pathway. Cancer Biomark. 2024;40:47–59.
Deng G, He J, Huang Q, Li T, Huang Z, Gao S, Xu J, Wang T, Di J. Ibrutinib inhibits BTK Signaling in Tumor-infiltrated B cells and amplifies Antitumor immunity by PD-1 checkpoint blockade for metastatic prostate Cancer. Cancers (Basel), 15 (2023).
Han HS, Jeong S, Kim H, Kim HD, Kim AR, Kwon M, Park SH, Woo CG, Kim HK, Lee KH, Seo SP, Kang HW, Kim WT, Kim WJ, Yun SJ, Shin EC. TOX-expressing terminally exhausted tumor-infiltrating CD8(+) T cells are reinvigorated by co-blockade of PD-1 and TIGIT in bladder cancer. Cancer Lett. 2021;499:137–47.
Freshour SL, Chen TH, Fisk B, Shen H, Mosior M, Skidmore ZL, Fronick C, Bolzenius JK, Griffith OL, Arora VK, Griffith M. Endothelial cells are a key target of IFN-g during response to combined PD-1/CTLA-4 ICB treatment in a mouse model of bladder cancer. iScience. 2023;26:107937.
Denis M, Grasselly C, Choffour PA, Wierinckx A, Mathe D, Chettab K, Tourette A, Talhi N, Bourguignon A, Birzele F, Kress E, Jordheim LP, Klein C, Matera EL, Dumontet C. Vivo syngeneic tumor models with Acquired Resistance to Anti-PD-1/PD-L1 therapies. Cancer Immunol Res. 2022;10:1013–27.
Zhao Z, Liu S, Sun R, Zhu W, Zhang Y, Liu T, Li T, Jiang N, Guo H, Yang R. The combination of oxaliplatin and anti-PD-1 inhibitor promotes immune cells infiltration and enhances anti-tumor effect of PD-1 blockade in bladder cancer. Front Immunol. 2023;14:1085476.
Li H, Chen J, Li Z, Chen M, Ou Z, Mo M, Wang R, Tong S, Liu P, Cai Z, Zhang C, Liu Z, Deng D, Liu J, Cheng C, Hu J, Zu X. S100A5 attenuates efficiency of Anti-PD-L1/PD-1 immunotherapy by inhibiting CD8(+) T cell-mediated anti-cancer immunity in bladder carcinoma. Adv Sci (Weinh). 2023;10:e2300110.
Okato A, Utsumi T, Ranieri M, Zheng X, Zhou M, Pereira LD, Chen T, Kita Y, Wu D, Hyun H, Lee H, Gdowski AS, Raupp JD, Clark-Garvey S, Manocha U, Chafitz A, Sherman F, Stephens J, Rose TL, Milowsky MI, Wobker SE, Serody JS, Damrauer JS, Wong KK, Kim WY. FGFR inhibition augments anti-PD-1 efficacy in murine FGFR3-mutant bladder cancer by abrogating immunosuppression. J Clin Invest, 134 (2024).
Tsuji S, Reil K, Nelson K, Proclivo VH, McGuire KL, Giacalone MJ. Intravesical VAX014 synergizes with PD-L1 blockade to enhance local and systemic control of bladder Cancer. Cancer Immunol Res. 2022;10:978–95.
Li L, Zeng X, Chao Z, Luo J, Guan W, Zhang Q, Ge Y, Wang Y, Xiong Z, Ma S, Zhou Q, Zhang J, Tian J, Horne D, Yuh B, Hu Z, Wei GH, Wang B, Zhang X, Lan P, Wang Z. Targeting alpha-ketoglutarate disruption overcomes Immunoevasion and improves PD-1 Blockade Immunotherapy in Renal Cell Carcinoma. Adv Sci (Weinh). 2023;10:e2301975.
Adachi Y, Kamiyama H, Ichikawa K, Fukushima S, Ozawa Y, Yamaguchi S, Goda S, Kimura T, Kodama K, Matsuki M, Miyano SW, Yokoi A, Kato Y, Funahashi Y. Inhibition of FGFR reactivates IFNgamma Signaling in Tumor cells to enhance the combined antitumor activity of Lenvatinib with Anti-PD-1 antibodies. Cancer Res. 2022;82:292–306.
Wu FTH, Xu P, Chow A, Man S, Kruger J, Khan KA, Paez-Ribes M, Pham E, Kerbel RS. Pre- and post-operative anti-PD-L1 plus anti-angiogenic therapies in mouse breast or renal cancer models of micro- or macro-metastatic disease. Br J Cancer. 2019;120:196–206.
Zelba H, Bedke J, Hennenlotter J, Mostbock S, Zettl M, Zichner T, Chandran A, Stenzl A, Rammensee HG, Gouttefangeas C. PD-1 and LAG-3 dominate checkpoint receptor-mediated T-cell inhibition in renal cell carcinoma. Cancer Immunol Res. 2019;7:1891–9.
Orlandella RM, Turbitt WJ, Gibson JT, Boi SK, Li P, Smith DL Jr., Norian LA. The antidiabetic Agent Acarbose improves Anti-PD-1 and rapamycin efficacy in preclinical renal Cancer. Cancers (Basel), 12 (2020).
Ferreiro-Pantin M, Anido-Herranz U, Betancor YZ, Cebey-Lopez V, Leon-Mateos L, Garcia-Gonzalez J, Garcia-Acuna SM, Fernandez-Diaz N, Tubio JMC, Lopez-Lopez R, Ruiz-Banobre J. Clinical, molecular, and immune correlates of the Immunotherapy response score in patients with advanced urothelial carcinoma under atezolizumab monotherapy: analysis of the phase II IMvigor210 trial. ESMO Open. 2023;8:101611.
Hopkins AM, Kichenadasse G, Karapetis CS, Rowland A, Sorich MJ. Concomitant Proton pump inhibitor use and survival in Urothelial Carcinoma Treated with Atezolizumab. Clin Cancer Res. 2020;26:5487–93.
Balar AV, Castellano DE, Grivas P, Vaughn DJ, Powles T, Vuky J, Fradet Y, Lee JL, Fong L, Vogelzang NJ, Climent MA, Necchi A, Petrylak DP, Plimack ER, Xu JZ, Imai K, Moreno BH, Bellmunt J, de Wit R. O’Donnell, Efficacy and safety of pembrolizumab in metastatic urothelial carcinoma: results from KEYNOTE-045 and KEYNOTE-052 after up to 5 years of follow-up. Ann Oncol. 2023;34:289–99.
Bellmunt J, de Wit R, Fradet Y, Climent MA, Petrylak DP, Lee JL, Fong L, Necchi A, Sternberg CN, O’Donnell PH, Powles T, Plimack ER, Bajorin DF, Balar AV, Castellano D, Choueiri TK, Culine S, Gerritsen W, Gurney H, Quinn DI, Vuky J, Vogelzang NJ, Cristescu R, Lunceford J, Saadatpour A, Loboda A, Ma J, Rajasagi M, Godwin JL, Homet Moreno B, Grivas P. Putative biomarkers of Clinical Benefit with Pembrolizumab in Advanced Urothelial Cancer: results from the KEYNOTE-045 and KEYNOTE-052 Landmark trials. Clin Cancer Res. 2022;28:2050–60.
Grimm MO, Esteban E, Barthelemy P, Schmidinger M, Busch J, Valderrama BP, Charnley N, Schmitz M, Schumacher U, Leucht K, Foller S, Baretton G, Duran I, de Velasco G, Priou F, Maroto P, Albiges L. .s. group, tailored immunotherapy approach with nivolumab with or without nivolumab plus ipilimumab as immunotherapeutic boost in patients with metastatic renal cell carcinoma (TITAN-RCC): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2023;24:1252–65.
Escudier B, Motzer RJ, Sharma P, Wagstaff J, Plimack ER, Hammers HJ, Donskov F, Gurney H, Sosman JA, Zalewski PG, Harmenberg U, McDermott DF, Choueiri TK, Richardet M, Tomita Y, Ravaud A, Doan J, Zhao H, Hardy H. George, Treatment Beyond Progression in patients with Advanced Renal Cell Carcinoma treated with Nivolumab in CheckMate 025. Eur Urol. 2017;72:368–76.
Albiges L, Tannir NM, Burotto M, McDermott D, Plimack ER, Barthelemy P, Porta C, Powles T, Donskov F, George S, Kollmannsberger CK, Gurney H, Grimm MO, Tomita Y, Castellano D, Rini BI, Choueiri TK, Leung D, Saggi SS, Lee CW, McHenry MB, Motzer RJ. First-line Nivolumab plus Ipilimumab Versus Sunitinib in patients without Nephrectomy and with an evaluable primary renal tumor in the CheckMate 214 Trial. Eur Urol. 2022;81:266–71.
Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ, Vaishampayan U, Berger R, Sezer A, Alanko T, de Wit R, Li C, Omlin A, Procopio G, Fukasawa S, Tabata KI, Park SH, Feyerabend S, Drake CG, Wu H, Qiu P, Kim J, Poehlein C. Bono, Pembrolizumab for treatment-refractory metastatic castration-resistant prostate Cancer: Multicohort, open-label phase II KEYNOTE-199 study. J Clin Oncol. 2020;38:395–405. de.
Peters S, Paz-Ares L, Herbst RS, Reck M. Addressing CPI resistance in NSCLC: targeting TAM receptors to modulate the tumor microenvironment and future prospects. J Immunother Cancer, 10 (2022).
Dong J, Miao J, Miao Y, Qu Z, Zhang S, Zhu P, Wiede F, Jassim BA, Bai Y, Nguyen Q, Lin J, Chen L, Tiganis T, Tao WA, Zhang ZY. Small molecule degraders of protein tyrosine phosphatase 1B and T-Cell protein tyrosine phosphatase for Cancer Immunotherapy. Angew Chem Int Ed Engl. 2023;62:e202303818.
O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103.
Ambrosini M, Rousseau B, Manca P, Artz O, Marabelle A, Andre T, Maddalena G, Mazzoli G, Intini R, Cohen R, Cercek A, Segal NH, Saltz L, Varghese AM, Yaeger R, Nusrat M, Goldberg Z, Ku GY, El Dika I, Margalit O, Grinshpun A, Murtaza Kasi P, Schilsky R, Lutfi A, Shacham-Shmueli E, Khan Afghan M, Weiss L, Westphalen CB, Conca V, Decker B, Randon G, Elez E, Fakih M, Schrock AB, Cremolini C, Jayachandran P, Overman MJ, Lonardi S, Pietrantonio F. Immune checkpoint inhibitors for POLE or POLD1 proofreading-deficient metastatic colorectal cancer. Ann Oncol. 2024;35:643–55.
Zhou Y, Li A, Yu H, Wang Y, Zhang X, Qiu H, Du W, Luo L, Fu S, Zhang L, Hong S. Neoadjuvant-adjuvant vs neoadjuvant-only PD-1 and PD-L1 inhibitors for patients with Resectable NSCLC: an Indirect Meta-Analysis. JAMA Netw Open. 2024;7:e241285.
Galle P, Finn RS, Mitchell CR, Ndirangu K, Ramji Z, Redhead GS, Pinato DJ. Treatment-emergent antidrug antibodies related to PD-1, PD-L1, or CTLA-4 inhibitors across tumor types: a systematic review. J Immunother Cancer, 12 (2024).
Rowe JH, Elia I, Shahid O, Gaudiano EF, Sifnugel NE, Johnson S, Reynolds AG, Fung ME, Joshi S, LaFleur MW, Park JS, Pauken KE, Rabinowitz JD, Freeman GJ, Haigis MC, Sharpe AH. Formate supplementation enhances Antitumor CD8 + T-cell fitness and efficacy of PD-1 blockade. Cancer Discov. 2023;13:2566–83.
Andreu-Saumell I, Rodriguez-Garcia A, Muhlgrabner V, Gimenez-Alejandre M, Marzal B, Castellsague J, Braso-Maristany F, Calderon H, Angelats L, Colell S, Nuding M, Soria-Castellano M, Barbao P, Prat A, Urbano-Ispizua A, Huppa JB, Guedan S. CAR affinity modulates the sensitivity of CAR-T cells to PD-1/PD-L1-mediated inhibition. Nat Commun. 2024;15:3552.
Fernandez-Barrena MG, Arechederra M, Colyn L, Berasain C, Avila MA. Epigenetics in hepatocellular carcinoma development and therapy: the tip of the iceberg. JHEP Rep. 2020;2:100167.
Deutsch JS, Lai J, Schenk KM, Soni A, Will EM, Engle LL, Xu H, Ogurtsova A, Madan V, Chong JK, Wang D, Green BF, Nguyen P, Schollenberger MD, Lipson EJ, Taube JM. Immune microenvironment of basal cell carcinoma and tumor regression following combined PD-1/LAG-3 blockade. J Immunother Cancer, 11 (2023).
Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV, Connolly RM, Santa-Maria CA, Wang J, Bahadur SW, Weickhardt A, Asch AS, Mallesara G, Clingan P, Dlugosz-Danecka M, Tomaszewska-Kiecana M, Pylypenko H, Hamad N, Kindler HL, Sumrow BJ, Kaminker P, Chen FZ, Zhang X, Shah K, Smith DH, De Costa A, Li J, Li H, Sun J, Moore PA. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. 2023;29:2814–24.
Mulholland M, Kritikou E, Katra P, Nilsson J, Bjorkbacka H, Lichtman AH, Rodriguez A, Engelbertsen D. LAG3 regulates T cell activation and plaque infiltration in atherosclerotic mice. JACC CardioOncol. 2022;4:635–45.
Kang JH, Zappasodi R. Modulating Treg stability to improve cancer immunotherapy. Trends Cancer. 2023;9:911–27.
Vesely MD, Zhang T, Chen L. Resistance mechanisms to Anti-PD Cancer Immunotherapy. Annu Rev Immunol. 2022;40:45–74.
Boyiadzis MM, Kirkwood JM, Marshall JL, et al. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. j Immunotherapy cancer. 2018;9:35.
Acknowledgements
We thank the generous support of Liaoning Cancer Hospital & Institute (Shenyang) and Dalian University of Technology(Dalian).
Funding
None.
Author information
Authors and Affiliations
Contributions
Original draft preparation, allocation, revision, and editing: Qiang Liu, Yujing Guan and Shenglong Li. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
None.
Consent for publication
None.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Following publication of the original article [1], the author reported that the published PDF version is incorrect as the information such as "(See Fig. 5)", "(Fig. 4; Table 3)", and "[170]" need to be removed and updated accordingly as shown in the correction article https://doi.org/10.1186/s12943-024-02121-9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Q., Guan, Y. & Li, S. Programmed death receptor (PD-)1/PD-ligand (L)1 in urological cancers : the “all-around warrior” in immunotherapy. Mol Cancer 23, 183 (2024). https://doi.org/10.1186/s12943-024-02095-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-02095-8