
Abstract
Ulcerative colitis (UC) is a challenging inflammatory gastrointestinal disorder, whose therapies encounter limitations in overcoming insufficient colonic retention and rapid systemic clearance. In this study, we report an innovative polymeric prodrug nanoformulation for targeted UC treatment through sustained 5-aminosalicylic acid (5-ASA) delivery. Amphiphilic polymer-based 13.5 nm micelles were engineered to incorporate azo-linked 5-ASA prodrug motifs, enabling cleavage via colonic azoreductases. In vitro, micelles exhibited excellent stability under gastric/intestinal conditions while demonstrating controlled 5-ASA release over 24 h in colonic fluids. Orally administered micelles revealed prolonged 24-h retention and a high accumulation within inflamed murine colonic tissue. At an approximately 60% dose reduction from those most advanced recent studies, the platform halted DSS colitis progression and outperformed standard 5-ASA therapy through a 77–97% suppression of inflammatory markers. Histological analysis confirmed intact colon morphology and restored barrier protein expression. This integrated prodrug nanoformulation addresses limitations in colon-targeted UC therapy through localized bioactivation and tailored pharmacokinetics, suggesting the potential of nanotechnology-guided precision delivery to transform disease management.
Similar content being viewed by others


Introduction
Oral drug administration, one of the most common routes for therapeutic delivery, is widely favored for its convenience, non-invasiveness, and patient compliance [1]. However, the oral route also presents several challenges that can impede the effectiveness of treatment [2]. The primary concerns is the variable bioavailability of drugs, influenced by factors such as gastrointestinal pH, enzymatic degradation. Furthermore, the first-pass metabolism in the liver can significantly alter the concentration of the drug located at the desired position, which lead to a higher dose to achieve the desired therapeutic effect, and in turn increase the risk of side effects [3]. Thus, taking full advantage of these characters is crucial in designing effective oral drug delivery systems, particularly when addressing chronic and localized conditions.
The advent of nanotechnology in drug delivery has revolutionized the field of oral medications [4]. Nanomedicines offer numerous advantages, including enhanced solubility, stability, and the potential for targeted delivery to specific sites within the body. However, despite these advancements, oral nanomedicines have their own set of challenges, such as premature release and robust release at target site [5]. Oral prodrug is another method also used to enhance the bioavailability and efficacy of active pharmaceutical ingredients [6]. Prodrugs are chemically modified, inactive derivatives of active drugs, designed to undergo transformation within the body to release the active drug. This strategy often addresses the limitations of direct drug delivery by improving solubility and stability. However, a major concern is ensuring that the conversion to the active drug occurs precisely at the targeted site [7]. In conditions where specific localization is required, achieving site-specific activation of prodrugs can be particularly challenging. Furthermore, the many enzymes in the gastrointestinal environment can activate the prodrug with robust kinetics, thus reducing the therapeutic efficiency. Thus, while oral prodrugs offer a promising avenue for improved drug delivery, addressing their inherent challenges is crucial for developing effective treatments, particularly for targeted therapy in diseases.
Ulcerative colitis (UC) is a chronic inflammatory bowel disease whose treatments primarily rely on oral-based small-molecule anti-inflammatory drugs, such as 5-aminosalicylic acid (5-ASA), aiming to induce and maintain remission and manage symptoms during flare-ups [8,9,10,11,12]. However, as an oral drug, its effectiveness for UC is often hampered by the challenges mentioned above [13,14,15]. Moreover, many patients with UC experience periods of increased intestinal motility and promoted emptying, which can further complicate the retention and effectiveness of orally administered therapies [16,17,18]. Thus, strategies to overcome retention and specificity would great benefit the therapeutic of UC.
Nanoparticles combining prodrugs have been used for 5-ASA oral delivery to treat UC [19,20,21,22,23,24,25]. These systems, designed to extend the drug’s presence in the colon, often involve surface modifications such as imparting a negative charge to target inflamed colonic mucosa selectively [26,27,28]. However, rapid drug release and subsequent clearance from the colon remain significant limitations of these systems, even with those who were linked through covalent bonds [29,30,31]. As a result, drugs get cleared before they have a chance to exert their therapeutic effect. Zero-order, sustained release allows constant therapeutic levels to be maintained, overwhelming drug-delivery systems with robust releasing [32,33,34,35,36,37]. Hence, the desired drug delivery nanoparticle designs should be equipped with not only targeted retention, but also extended slow-release profile.
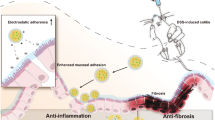
Our laboratory has developed a series of polymers for different applications [38,39,40,41,42,43,44,45]. Very interestingly, we found that the molecules trapped inside the hyperbranched structure of polymer can alternate its reaction kinetic towards the outside environment [46]. Thus, in this study our investigative efforts focused on the development of a polymeric prodrug nanoformulation, characterized by its structural simplicity, which affords targeted and sustained release of therapeutic agents within the colon (Fig. 1a). This nanoformulation prodrug design involves an amphiphilic polymer-based micellar architecture, yielding a delivery vehicle of nanosized micelles to avoid premature drug absorption. The surface charge of these micelles has been engineered to possess a negative charge, thereby enhancing affinity for the positively charged inflamed colonic tissue of UC and ensuring preferential localization [47]. 5-ASA is conjugated via an azo linkage, which is specifically cleavable within the colonic environment by azoreductase enzymes [48,49,50,51]. The polymer’s side chains strategically hinder the prodrug, moderating its release and enabling a controlled, extended drug release profile. This design effectively tempers the rapid cleavage of azo-linked molecules, allowing for an extended drug release timeline that surpasses previous research in both retention and drug release duration. Such a desired drug delivery behavior led to a decrease of 5-ASA dosage dramatically but with a commendable therapeutic effect, offering a novel drug delivery system that promises to improve patient outcomes by addressing the limitations of conventional treatments.

a Proposed mechanism of P3 to treat UC. b Temporal dependence of UV–Vis absorption profile for P3 (0.5 mg \(\hbox {mL}^{-1}\)) in PBS at \(25 ^{\circ }\)C. c Statistical distributions of the size for micelles formed by P3 (0.5 mg \(\hbox {mL}^{-1}\)) determined by DLS in PBS at \(25 ^{\circ }\)C. d TEM image of micelles formed by P3 in PBS. e Temporal dependence of size for P3 (0.5 mg mL^-1) in PBS at \(25 ^{\circ }\)C. f Plots of the emission intensity (\(\lambda _{Ex}\) = 494 nm, \(\lambda _{Em}\) = 512 nm), recorded at \(25 ^{\circ }\)C after combining MeOH solutions of BODIPY and P3, distilling the solvent off under reduced pressure, and dispersing the residue in PBS (1 mL), against the polymer concentration. g Zeta-Potential of P2 and P3 (0.5 mg \(\hbox {mL}^{-1}\)) in PBS at \(25 ^{\circ }\)C.
Results and discussion
Synthesis and characterization
The synthesis of the 5-ASA polymeric prodrug (P3) involved the incorporation of three essential components: a hydrophobic poly(methacrylate) backbone, hydrophilic side chain, and azo-linked 5-ASA prodrug units. This was achieved through a three-step procedure, as described in Scheme S1. In the first step, a radical copolymerization reaction was carried out using polyethylene glycol methacrylate (PEGMA, Mn = 950 g \(\hbox {mol}^{-1}\)) and N-Boc-protected N-(2-aminoethyl)methacrylamide as monomers, with azobisisobutyronitrile (AIBN) serving as the initiator. The reaction took place in tetrahydrofuran at \(75 ^{\circ }\textrm{C}\) for 2 days. The integration of the proton signals from the PEG side chain’s terminal methyl group (-\(\hbox {OCH}_3\), 3.4 ppm) and the Boc protecting group (1.5 ppm) in the \({}^1\)H NMR spectrum (Figure S14) allowed for an estimation of the mole ratio of the two monomers, which was found to be 1:3. The spectrum also indicated the complete elimination of the monomers, as the olefin signals between 5–6 ppm disappeared entirely. Subsequently, the Boc-protecting groups were cleaved using HCl (4 M in dioxane), resulting in the formation of a cationic copolymer P2. Finally, the azo-linked 5-ASA prodrug unit was conjugated to the polymer backbone by reacting the primary amines with compound 3 at a 1:2 molar ratio, employing \(\hbox {NaBH}_3\)CN as a reducing agent. The absence of peaks around 10.1 ppm in the NMR spectrum of P3 (Figure S17), indicating the proton signal from an aldehyde group, demonstrates the covalent linkage of the prodrug unit to the polymer. GPC data (Figure S18) showed a monodispersed molecular weight distribution of P1, P2 and P3, with their Mw calculated as 22.5, 17.1 and 18.8 kDa, respectively.
The UV–vis absorption measurements demonstrated the presence of an absorption peak at 370 nm in P3, which was identical to the peak observed in compound 3 (Fig. 1b and S1b). By utilizing UV absorption measurements and the molar extinction coefficient of compound 3 (22,800 \(\hbox {M}^{-1}\) \(\hbox {cm}^{-1}\), calculated from Figure S1c), it was determined that the loading of 5-ASA in the polymer was 11.5 wt%. The consistent absorption profile of P3 over a period of 72 h provided evidence of the stability of the azo bond under physiological conditions. P3 effectively self-assembled into micelles with an average diameter of approximately 13.5 nm, with a PDI of 0.43, as depicted in Fig. 1c, displaying a nearly uniform size distribution. The size of the micelles was further validated by TEM image (Fig. 1d). These micelles exhibited exceptional stability, with no significant change in size observed over a 72-hour storage period at room temperature in PBS (Fig. 1e). It is crucial to note that the large size of the micelles, in comparison to the small molecule 5-ASA (which is rapidly cleared), is expected to ensure prolonged retention in the inflamed colon. As demonstrated in Fig. 1f, the critical micelle concentration (CMC) was determined to be 0.59 \(\mu\)M, preventing micelle disassembly upon dilution in biological fluids. The inclusion of anionic azo prodrug units within the micelles altered the zeta potential from +19 mV for the precursor P2 to \(-\)24.2 mV for P3 (Fig. 1g). This negative surface charge is anticipated to minimize adhesion in the healthy colon, where the mucus layer also carries a negative charge. Conversely, the negative charge facilitates adhesion to the positively charged inflamed colonic epithelium [52]. These favorable physicochemical properties of the 5-ASA polymeric prodrug offer promising prospects for targeted and sustained delivery of 5-ASA for colitis therapy.
In vitro drug release
The release profile of 5-ASA from a model azo prodrug was first investigated in vitro using compound 3. Compound 3 contains an azo linker attaching a 4-aminobenzaldehyde group to 5-ASA, providing a representative small molecule mimic of the polymeric azo prodrug. Analysis of compound 3 by HPLC revealed a peak at a retention time of 4.2 min under the elution condition (Fig. 2a). To simulate colonic azoreductase activity, compound 3 (100 \(\mu\)M) was incubated with the reducing agent sodium dithionite (SDT) in double distilled water [53,54,55]. At an SDT concentration of 2.5 mM, the peak at 4.2 min disappeared within 30 min of incubation at room temperature (Fig. 2a). Concurrent formation of a new peak at 1.1 min was observed, corresponding to 5-ASA based on comparison with the 5-ASA standard, released after the reduction of the azo bond.
Similar release behavior was observed for polymeric prodrug P3. The original absorption peak at 370 nm decreased upon treatment with different equivalents of SDT in double distilled water for 0.5 h (Fig. 2b). Direct MS analysis of the reaction mixture showed the appearance of a peak at m/z 152.1 after 0.5 h, confirming 5-ASA release (Fig. S2). In simulated gastric fluid (SGF) and simulated intestinal fluid (SIF), P3 micelles remained intact, and no 5-ASA was detected over 3 h by MS (Fig. S2). DLS measurements were consistent with the spectrophotometric and MS data. The mean diameter of P3 micelles remained around 12 nm after incubation in SGF, while 10 nm in SIF, for 24 h each. In contrast, a rapid decrease in size to 5 nm was observed within the first 4 h incubation in SIF containing 25 eq SDT.

a HPLC chromatograms of model compound 3 (0.1 mM) after reaction with SDT (2.5 mM) for 0.5 h. C18 chromatography performed using 10–100% acetonitrile: water gradient mobile phase. b UV–vis absorption spectra of P3 (0.05 mg \(\hbox {mL}^{-1}\)) treated with varying SDT equivalents. c Size distribution by DLS for P3 micelles after incubation in SGF, SIF, or SIF with SDT. Results are the average of 3 measurements. HPLC chromatograms of P3 after incubation in d SI contents and e colon contents. SI contents and colon contents were collected from healthy mice. P3 (3 mg \(\hbox {mL}^{-1}\)) were incubated with SI contents for 2, 8 and 12 h, or with colon contents for 2, 4, 8, 12 and 24 h, followed by ultrafiltration to remove polymers and proteins. Size exclusion chromatography was performed using 150 mM PBS as mobile phase. f Accumulated release of 5-ASA over time from P3 in gastric, small intestinal and colonic conditions. The blue area stands for the 5-ASA released after 2 h. Equivalent data for SSZ highlighting release in g SI, h colonic contents, and i accumulative release of 5-ASA over time from SSZ in gastric, small intestinal and colonic conditions. Size exclusion HPLC used MeOH: PBS (1:1, v: v). Control samples include 5-ASA standard (1 mM), SI contents only, colon contents only, and SSZ standard (1 mM).
After the confirmation of the 5-ASA release mechanism, the in vitro release kinetics from polymeric prodrug P3 versus small molecule sulfasalazine (SSZ), a commercially available azo-based prodrug of 5-ASA, were investigated by HPLC. P3 and SSZ were firstly incubated in gastric contents for 4 h and small intestinal contents for 12 h, representing typical maximum transit times in mice, [56,57,58,59] before injected into the size exclusive column. HPLC analysis of P3 conducted under gastric contents and small intestinal contents did not reveal any detectable presence of 5-ASA, as exemplified in Figures S3a and 2d, indicating the excellent stability of P3 in these environments. In contrast, SSZ exhibited no 5-ASA release in gastric contents (Figure S3b) while a partial release under intestinal conditions, with a small peak emerging at the retention time of standard 5-ASA indicating approximately 25% release in 12 h (Figs. 2g and S4a). This release behavior in the small intestine decreases its potential therapeutic effect.
The transformation occurred when P3 was exposed to the colon contents, where the presence of azoreductase activity triggered the selective release of 5-ASA. HPLC quantification of the released 5-ASA over a 24-hour incubation period, representing typical transit time, [60,61,62] unveiled a controlled and sustained release profile, with approximately 60.0% of the loaded 5-ASA being liberated at a sustained rate(Figs. 2e and f). In stark contrast, SSZ exhibited a significantly different release profile. Within the colon, SSZ released the majority of its 5-ASA payload (82.9%) within a mere 2-hour period and minimal further release beyond 2 h (Figs. 2h, i and S4b), as opposed to the controlled and sustained release observed with P3. Such a robust release profile may not guarantee the maintenance of effective 5-ASA concentrations afterward, potentially resulting in a diminished therapeutic effect. The area under the curve for 5-ASA concentration over time after 2 h was 6.5-fold higher for P3 compared to SSZ, indicating substantially enhanced long-term drug exposure. P3’s sustained release kinetics closely simulate the colonic environment, confirming specificity of sustained 5-ASA release in response to azoreductase. Compared to SSZ’s initial burst release, P3’s sustained release profile enables maintenance of therapeutic 5-ASA concentrations in inflamed colonic tissue for a prolonged duration. Such data prove that our novel drug delivery system presents a promising approach to controlled delivery to optimize ulcerative colitis therapy, addressing the limitations associated with conventional treatments with small molecules, like SSZ.
Retention in colon
Prior to the in vivo test, the toxicity of P2 and P3 was first evaluated in vitro using Caco-2 cells and RAW 264.7 macrophages (Figure S5). Cells were incubated with varying concentrations of polymers P2 and P3 for 24 h before assessing cell viability using the CCK-8 assay. Both polymers exhibited minimal toxicity in either cell line up to the maximum tested concentration of 5 mg \(\hbox {mL}^{-1}\). It demonstrates that the polymers exhibit excellent biocompatibility and negligible cytotoxicity in colon-relevant cell types at concentrations well above the intended therapeutic range.
To further validate the electrostatic targeting capabilities of the polymeric prodrug nanoparticles, in vitro adhesion studies were performed using surfaces coated with mucin, a major component of colonic mucus. Freshly cleaved mica sheets were coated with a 1 mg \(\hbox {mL}^{-1}\) mucin solution to create an anionic surface mimicking healthy colonic mucosa, then incubated with TRITC-labeled P3 (TP3, absorption and emission spectra presented in Figure S6) nanoparticles (1 mg \(\hbox {mL}^{-1}\)). Fluorescence microscopy revealed minimal nanoparticle adhesion, with sparse coverage on the mucin layer (Fig. 3b). This aligns with the goal of avoiding retention in unaffected colon. In contrast, when the mucin coatings were treated with the cationic polyetherimide (1 mg\(\hbox {mL}^{-1}\)) to generate a net positive surface charge, TP3 nanoparticles exhibited an dramatic increase in adhesion density. As similar charge reversal is observed in UC lesions compared to healthy mucosa, this design could ensure an enhanced adherence to the inflammation part [52]. Taken together, these results confirm that the anionic polymeric prodrug nanoparticles can selectively bind to and be retained by positively charged inflamed colonic tissue versus the negatively charged healthy mucosa.
In vivo evaluation of P3 and a small molecule dye, Nile Blue (NB), was performed to assess their respective retention profiles in healthy and DSS-induced colitic mice using comprehensive whole-body fluorescence imaging (as depicted in Fig. 3c). Due to the fluorescent quenching effect of azo compound towards most fluorophores, TP3 is not suitable for whole body imaging in this case [63]. Thus we applied a NIR BODIPY (NBP, structure and spectra shown in Figure S1 and S7) to be encapsulated in P3 micelle for tracking [64]. The investigation commenced immediately following oral gavage and entailed tracking at time points of 0.5, 3, 6, 12 and 24 h post-administration. At 0.5 h, both P3 encapsulating NBP (NBP@P3) and NB exhibited pronounced signals within the upper bally region, indicative of their initial gastric retention. By the 3-hour mark, NB fluorescence in DSS-induced colitic mice exhibited a rapid decline, primarily attributed to its swift evacuation from the gastrointestinal tract, while in healthy mice, residual signals had a moderate decrease. As the evaluation continued, the 6-hour time point revealed a remarkable reduction of NB fluorescence by approximately 77% in healthy mice, attributable to processes involving absorption and subsequent systemic clearance. In contrast, within the DSS-induced colitic mice, NB fluorescence became nearly undetectable, aligning with the notable lack of retention.

a Proposed mechanism of inflamed colon-targeted delivery for P3. The negative surface charge minimizes mucoadhesion in healthy colon. Electrostatic adhesion to positively charged inflamed colonic tissue enables site-specific retention. Sustained colonic release improves local efficacy. b Evaluation of polymeric prodrug nanoparticle adhesion to surfaces with defined charge. Fluorescence microscopy images showing minimal adhesion of TP3 nanoparticles on mucin protein coatings while increased TP3 adhesion when mucin coatings are treated with the cationic polymer polyetherimide to provide a positively charged surface. Scale bar: 200 \(\mu\)m. c Whole body fluorescence images at 0.5, 3, 6, 12, and 24 h after oral gavage in healthy and DSS colitic mice, with background signal subtracted. NBP@P3 exhibits prolonged retention up to 24 h in DSS mice compared to rapid clearance of NB. d, e Quantitative analysis of fluorescence intensity in the belly region from whole-body images in Figure 3c. f NB biodistribution at 3 h shows fluorescence appeared in the livers and low levels in other organs. g Biodistribution of NBP@P3 fluorescence intensity in excised organs at 3 h. No fluorescence is detected in the heart, liver, spleen, lung, or kidneys. h NBP@P3 fluorescent image in the isolated colonic region of healthy versus DSS colitic mice over 24 h. i Quantitative analysis of fluorescence intensity in the excisional colonic region of healthy versus DSS colitic mice after treating with NBP@P3 for 24 h
In sharp contrast to the behavior of NB, NBP@P3 demonstrated a strikingly different retention profile (Fig. 3c–e). At the 6-hour interval, NBP@P3 exhibited intense fluorescence specifically within the lower belly region of DSS-induced colitic mice. This observation underscores the exceptional ability of the polymeric prodrug to achieve prolonged retention and targeted delivery to inflamed colonic tissue. Subsequent measurements at the 12-hour time point unveiled a distinctive outcome: NB fluorescence remained undetectable in both healthy and DSS-induced colitic mice, signifying complete clearance from the system. In contrast, NBP@P3 fluorescence, while exhibiting a reduction compared to the 6-hour signals, remained readily detectable within the colons of both groups. Ultimately, at the 24-hour time point, NBP@P3 fluorescence persisted at detectable levels in DSS-induced colitic mice, further reinforcing its capacity for prolonged retention and targeted delivery to inflamed colonic tissue. In contrast, within the healthy mice, NBP@P3 exhibited a comparable reduction in fluorescence levels, highlighting the similarities in their clearance rates under different physiological conditions. Considering the quicker clearance rates in colitic mice, P3 demonstrated a distinct advantage in terms of retention [65].
Fluorescence imaging of excised organs unveiled distinct biodistribution patterns, providing further insights into the exceptional performance of NBP@P3 in targeted colitis therapy. In contrast to NB-treated mice, where fluorescence signals primarily localized in the livers at the 3-hour mark (Fig. 3f), NBP@P3 exhibited an entirely different distribution profile. Notably, NBP@P3 displayed negligible fluorescence in critical organs such as the heart, liver, spleen, and kidneys (Fig. 3g). The nanoparticle formulation NBP@P3 exhibited remarkable retention and colon-targeting capabilities, evidenced by significantly higher fluorescence intensities observed in the colons of DSS-induced colitic mice compared to their healthy counterparts at the 24-hour time point (Fig. 3h and 3i). This preferential accumulation can be attributed to the electrostatic interactions facilitated by the anionic surface charge of NBP@P3. Notably, the accelerated gastrointestinal transit and emptying typically observed in UC mice would be expected to reduce the retention time of orally administered agents. However, the enhanced retention of NBP@P3 in the DSS-induced colitic mice suggests a prolonged localization within the inflamed colonic tissue. Such enhanced retention is attributed to the electrostatic interactions arising from NBP@P3’s negative surface charge, facilitating its specific adhesion to the positively charged colonic mucosa in the context of colitis.
Therapeutic efficacy
The therapeutic efficacy of P3 was evaluated in vivo using a DSS-induced mouse model of colitis, [66] along with controls including precursor polymer P2 and a physical mixture of P2 + free 5-ASA. Mice were provided 2.5% DSS in drinking water for 7 days to induce colonic inflammation (Fig. 4a). They were then treated by daily oral gavage for 7 days with either PBS, 5-ASA (30 mg \(\hbox {kg}^{-1}\)), P2 (270 mg \(\hbox {kg}^{-1}\)), P2 + 5-ASA (270 mg \(\hbox {kg}^{-1}\) + 30 mg \(\hbox {kg}^{-1}\)), or an equivalent 5-ASA dose of P3 (300 mg \(\hbox {kg}^{-1}\), equivalent to 30 mg \(\hbox {kg}^{-1}\) 5-ASA). During the 7-day treatment period, P3-treated mice halted weight loss and began gaining starting on day 9 (the second day post-treatment), while other groups continued declining until day 11 (Fig. 4b). After the 7-day treatment, P3-treated mice weighed only 8.34% less than healthy controls with no significant difference (Fig. 4c). In contrast, PBS, 5-ASA, P2, and P2+5-ASA groups lost 21.06% (P < 0.0001), 17.09% (P < 0.0001), 22.63% (P < 0.0001), and 17.43% (P < 0.0001) of the body weight respectively compared to controls. These data demonstrate P3’s excellent efficacy in ameliorating DSS-induced weight loss. The polymer backbone P2 alone showed no therapeutic effect, confirming that efficacy arises from targeted 5-ASA delivery rather than the carrier polymer.
A similar trend was seen in changes of the Disease Activity Index (DAI), an integration of weight loss, stool consistency, and fecal blood [67, 68]. In PBS and P2 groups, DAI peaked at 10±1.7 and 10±1 on days 8–9 before recovering slightly (Fig. 4d). However, DAI remained elevated at 5.67±0.94 and 5.87±1.36 on day 14 (both p < 0.0001 vs. control) (Fig. 4e). 5-ASA and P2+5-ASA groups followed similar trends, peaking at day 8-9 and dropping to 4.88±1.05 and 5.13±1.05 by day 14 (p < 0.0001 vs. control). In contrast, the DAI of the P3 treatment group began to decline on day 9 of the experiment, showing an earlier recovery, and dropped to 2.25±1.09 on the fourteenth day of the experiment, which was significantly lower than that of the other groups (p < 0.01). By integrating multiple metrics of disease progression, the DAI results further confirm the enhanced therapeutic efficacy of P3.
In addition to reduced weight loss, P3 treatment markedly attenuated DSS-induced colonic shortening, a marker of inflammation and tissue damage (Fig. 4f and g) [69]. On day 14, the average colon length of PBS and P2-treated mice were 6.23 cm and 6.38 cm respectively, compared to 7.87 cm for healthy controls. 5-ASA and P2+5-ASA partially preserved colon length to 6.43 cm and 6.54 cm respectively (P < 0.0001 vs. control), while P3-treatment resulted in a length of 7.35 cm (P < 0.05 vs. control), very close to healthy mice. The spleen/body weight ratio was also calculated as a metric of systemic inflammation. In healthy mice, the average spleen/body weight ratio was 27.04 (Fig. 4h). This was significantly increased to 83.27 in PBS-treated colitic mice, indicative of splenomegaly. Treatment with standard 5-ASA moderately reduced this ratio to 53.35, while the polymeric prodrug P3 decreased it back to 40.65, similar to that of healthy mice. Mice receiving the precursor polymer P2 alone without 5-ASA conjugation exhibited a ratio of 76.73, no different than PBS controls. The combination of P2+5-ASA resulted in an intermediate ratio of 57.38, comparable to 5-ASA-treated mice but higher than P3-treated ones.

Therapeutic efficacy of P3 compared to controls against DSS-colitis in mice. a Schematic of the experimental design. Mice were randomized into 6 groups (Control: PBS+normal water, PBS+DSS water, 5-ASA+DSS water, P2+DSS water, P2+5-ASA+ DSS water, P3+DSS water). Colitis was induced with 2.5% DSS drinking water for 7 days followed by 7-day treatment with oral gavage of PBS, 5-ASA, P2, P2+5-ASA, or equivalent 5-ASA dose of P3. b Body weight changes from day 1 to day 14 and c the statistical chart of weight change on day 14. d DAI changes from day 0 to day 14 and e the statistical chart of the disease index on day 14. f Schematic diagram of colonic tissue in each group and g the statistical chart Colon length on the day 14. h Spleen/Body weight ratio. i IL-1\(\beta\), j IL-6 and k TNF-\(\alpha\) in serum. All data represent mean ± SEM, n = 3 mice per group. Statistical significance was determined by Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to healthy control or as indicated
Similarly, serum levels of pro-inflammatory cytokines IL-1\(\beta\), IL-6, and TNF-\(\alpha\) were massively overexpressed in PBS-treated colitic mice compared to healthy controls (Fig. 4i–k) [70, 71]. 5-ASA partially reduced their levels by 64%, 40%, and 51% respectively, relative to PBS-treated mice. However, P3 decreased IL-1\(\beta\) by 97%, IL-6 by 77%, and TNF-\(\alpha\) by 89% back to basal levels, a far greater suppression compared to 5-ASA. It is important to note that the levels of IL-1\(\beta\), IL-6, and TNF-\(\alpha\) in P3 treated mice showed no significant difference compared to the healthy mice. Mice receiving P2 alone showed moderate cytokine changes compared to PBS. The combination P2+5-ASA treatment lowered cytokines to a degree similar to 5-ASA alone but was significantly higher than P3-treated mice.
These results suggest that the conjugation of 5-ASA to the polymeric carrier P3 as an azoreductase-activated prodrug resulted in the greatest suppression of systemic and intestinal inflammatory markers as well as superior therapeutic efficacy in DSS-induced colitis. The polymer alone did not confer bioactivity, and the physical combination with 5-ASA had no enhancement compared to 5-ASA alone, highlighting the unique benefits of the prodrug nanoformulation. It is noteworthy to mention that this platform utilized a notably reduced dosage of 5-ASA at 30 mg \(\hbox {kg}^{-1}\), 60% lower than the dosages in recent research cases, yet achieved commendable therapeutic effects. These results demonstrate the potential of this polymeric prodrug approach to improve colitis treatment through targeted and sustained 5-ASA delivery to the inflamed colonic mucosa.
Histological analysis
H&E staining was performed on colon sections to assess tissue morphology and inflammation (Fig. 5 and S8-S10). Healthy control mice displayed intact colonic epithelium with abundant goblet cells and no immune cell infiltration. Mice receiving only DSS showed extensive disruption of epithelial architecture, massive loss of goblet cells, submucosal edema, and massive infiltration of immune cells including neutrophils and macrophages. Mice treated with just the precursor polymer P2 after DSS induction showed similar histological features as the DSS alone group, indicating the polymer carrier itself did not confer bioactivity. DSS mice receiving 5-ASA therapy displayed moderate improvement in tissue integrity compared to DSS controls, with some residual goblet cell loss and immune cell infiltration. The combination therapy of P2+5-ASA resulted in an intermediate degree of structural damage and inflammation. In contrast, mice treated with P3 showed remarkably intact colonic morphology comparable to healthy controls, with an intact epithelial barrier, abundant goblet cells, and minimal immune cell infiltration. P3 therapy effectively preserved the colonic epithelial architecture and suppressed inflammation, highlighting its significant therapeutic efficacy compared to standard 5-ASA treatment.
To further evaluate the efficacy of polymeric prodrug P3 at a molecular level, immunofluorescence staining was performed to visualize the expression and localization of key tight junction proteins that maintain colonic epithelial barrier integrity (Figs. 5b–d, S7-S9). These included ZO-1, Occludin, and Claudin-1 in colon sections from healthy mice or DSS-colitic mice treated with various regimens described previously. Confocal microscopy revealed intense staining and continuous membrane localization of all three proteins in a healthy control colon. In contrast, mice with untreated colitis showed severely disrupted staining and gaps between epithelial cells, indicating loss of barrier function. Notably, mice receiving P3 therapy displayed a remarkable restoration of tight junction protein expression, organization, and localization profiles matching healthy control. In contrast, only partial recovery was observed after treatment with standard 5-ASA or P2+5-ASA combination, while precursor polymer P2 alone showed no improvement versus untreated colitic mice. Taken together with the H&E analysis, these results confirm that sustained targeted delivery of 5-ASA mediated by the polymeric prodrug nanoformulation effectively preserves colonic barrier integrity on histological and molecular levels in this UC model. The ability to restore tight junction protein function highlights its potential to improve clinical outcomes.

Representative micrographs and quantitative analysis a H&E, b Occludin, c Claudin-1 and d ZO-1 stained colon sections from mice in the various treatment groups. After the 7-day treatment period, colon samples were harvested, fixed in 42% paraformaldehyde, and embedded in paraffin. Sections were cut to 4 \(\mu\)m thickness and stained. Images were acquired on an upright microscope for H&E staining and rest using a confocal microscope. P3-treated mice showed preserved colonic morphology similar to healthy controls, while other DSS groups exhibited damage that was partially ameliorated by 5-ASA treatment. Scale bar: 300 \(\mu\)m
Biocompatibility
To evaluate the potential toxicity of the lead polymeric prodrug P3, major organs including the heart, liver, spleen, lungs, and kidney were harvested from colitic mice treated with P3 and compared to controls (Figure S11). Microscopic examination of H &E stained tissue sections showed no observable differences in morphology or presence of pathologic signs between P3-treated and control mice in any of the analyzed organs. Heart sections displayed normal cardiomyocyte architecture with no inflammatory cell infiltration. Hepatocytes, splenocytes, pulmonary alveoli, renal tubules, and all appeared intact without necrosis or apoptosis. Collectively, these results demonstrate that systemic administration of polymeric prodrug P3 does not induce toxicity or physiologic changes in major organs. The favorable safety profile and biocompatibility of P3 support its potential translation for colitis therapy.
Methods
Chemicals were purchased from commercial sources and used as received. \(\hbox {H}_2\)O (18.2 M\(\Omega\)-cm) was purified with a Barnstead International NANOpure DIamond Analytical system. NMR spectra were recorded with a Zhongke Niujin QUANTUM-I-400MHz. GPC was performed with a Phenomenex Phenogel 5-\(\mu\)m MXM column (7.8 \(\times\) 300 mm) operated with a Malvern TDA-305 system in THF at a flow rate of 0.7 mL \(\hbox {min}^{-1}\) . Monodisperse polystyrene standards (5,000 - 123,000) were employed to determine the Mn of the polymers from the GPC traces. Transmission electron microscopy (TEM) was characterized with FEI Talos F200S. DLS measurements were performed with a Malvern Zetasizer Nano ZS ZEN3600 apparatus. Absorption and emission spectra were recorded with a ThermoFisher Varioskan LUX. High-performance liquid chromatography (HPLC) was performed with an Agilent Poroshell 300SB-C18 column (2.1 \(\times\)75 mm) or Agilent AdvanceBio SEC 300A column (2.6 \(\times\)150 mm) operated with an Agilent 1260 infinity II LC system. Fluorescence images were recorded with a Nikon A1 confocal laser-scanning microscope.
Synthesis of 3
4-Aminobenzaldehyde and sodium nitrite were dissolved in 50 mL of water in a molar ratio of 1:1.3. To this mixture, concentrated hydrochloric acid in a molar ratio of 1:4 was added. The reaction mixture was stirred at \(4 ^{\circ }\textrm{C}\) in an ice bath for 30 min. Subsequently, a solution of sodium hydroxide in salicylic acid was added to adjust the pH to 12 using 1 M NaOH solution, followed by stirring in an ice bath for another 3 h. Upon completion of the reaction, the reaction mixture was filtered and dried, yielding compound 3 as a brownish solid with a yield of 80%. \({}^1\)H NMR (400 MHz, DMSO-d6, \(\sigma\)): 10.11 (s, 1 H), 8.39 (d, J = 2.4 Hz, 1 H), 8.12 (d, J = 8.1 Hz, 3 H), 8.04 (d, J = 8.2 Hz, 2 H), 7.19 (d, J = 8.9 Hz, 1 H); \({}^{13}\)C NMR (400 MHz, DMSO-d6, \(\sigma\)): 193.10, 171.23, 171.12, 156.32, 142.93, 136.75, 131.28, 127.61, 127.56, 122.90, 119.65, 118.80; HRMS (ESI) m/z: [M - H]\({}^-\) calcd for C14H9N2O4, 269.0568; found, 269.0580.
Synthesis of P1
Polyethylene glycol methyl ether methacrylate (PEGMA, Mn = 950 g \(\hbox {mol}^{-1}\)) and Boc-protected N-(2-aminoethyl)methacrylamide were dissolved in anhydrous tetrahydrofuran (THF) in a molar ratio of 1:3. To this mixture, azobisisobutyronitrile (AIBN) at 3 mol% relative to the monomers was added as the initiator. The reaction was conducted at \(75 ^{\circ }\textrm{C}\) in an oil bath under a nitrogen atmosphere for 48 h. Afterward, THF was removed from the reaction mixture by vacuum distillation. The polymer was purified by LH-20 gel column chromatography (methanol/dichloromethane = 1:1, v/v) to obtain P1 as a colorless, transparent, viscous liquid with a yield of 58%. \({}^1\)H NMR (400 MHz, \(\hbox {CDCl}_3\)\(\sigma\)): 3.80 - 3.56 (m, 83 H), 3.41 (s, 3 H), 3.36 - 3.09 (m, 7 H), 2.06 - 1.91 (m, 23 H), 1.55–1.40 (m, 24 H).
Synthesis of P2
Polymer P1 was stirred in a 4 M solution of HCl in dioxane at room temperature for 2 h. Subsequently, dioxane and HCl were removed from the reaction mixture by vacuum distillation. The resulting polymer was then dried overnight in a vacuum oven to obtain P2 as a colorless, transparent, viscous liquid with a yield of 99%. \({}^1\)H NMR (400 MHz, CDCl3, \(\sigma\)): 3.80 - 3.55 (m, 82 H), 3.50–3.10 (m, 19 H), 1.41–0.73 (m, 8 H).
Synthesis of P3
Polymer P2 and compound 3 were dissolved in methanol at a molar ratio of 1 (amount of amine): 2. After stirring at room temperature for 1 h, a molar equivalent of sodium cyanoborohydride was added, and the mixture was stirred overnight at room temperature. The methanol in the reaction mixture was then removed by vacuum distillation, and the resulting product was purified using LH-20 gel column chromatography (methanol/dichloromethane = 1:1, v/v). The purified polymer was designated as P3 and appeared as a yellow solid with a yield of 73.6%. \({}^1\)H NMR (400 MHz, \(\hbox {D}_2\)O, \(\sigma\)): 9.02–6.05 (m, 3 H), 4.50–3.36 (m, 67 H), 3.34–3.25 (m, 3 H), 1.70–0.95 (m, 5 H), 0.95–0.30 (m, 2 H).
Synthesis of TP3
P3 (50 mg) was dissolved in 5 mL of potassium carbonate buffer (pH 9). TRITC was dissolved in DMSO at 20 mM and added to the polymer solution. The reaction was protected from light and allowed to proceed overnight at room temperature with stirring. The labeled polymer was purified by LH-20 gel column chromatography (methanol/dichloromethane = 1:1, v/v) to obtain TP3 as a yellow solid.
Synthesis of NBP. The synthesis of NBP followed a previous literature [64]. In the presence of piperidine (1.0 mL) and acetic acid (1.0 mL), a solution of BODIPY (1 mmol) and p-tolualdehyde (3 mmol) in toluene (15 mL) was heated at reflux by using a three-necked flask equipped with a Dean-Stark apparatus. The resulting residue was purified by column chromatography on neutral silica gel (hexane/EtOAc = 4 : 1) to give NBP in 76% yield (0.76 mmol).
Preparation of nanoparticle dispersions
A suitable volume of methanol solutions containing P2, P3 or TP3 (100 mg \(\hbox {mL}^{-1}\)) was transferred to a glass vial. The solvent was distilled off under reduced pressure, the residue was dispersed in PBS (sterile, 1 mL) and the mixture was sonicated for 5 min. The dispersion was then used for the spectroscopic and cellular experiments without further purification.
TEM
0.2 mg \(\hbox {mL}^{-1}\)of micelles suspension was diluted 5 times with \(\hbox {H}_2\)O and then dropped onto copper mesh. Then, ammonium molybdate was used for staining for 30 s and dried overnight at \(75 ^{\circ }\textrm{C}\). The samples were then imaged with FEI Talos F200S.
DLS and Zeta potential
0.5 mg \(\hbox {mL}^{-1}\)the nanoparticle formulation of P2 or P3 was prepared as described earlier. Subsequently, DLS and zeta potential measurements were performed using a Malvern Zetasizer Nano ZS ZEN3600 apparatus. The measurements were conducted in triplicate to ensure accuracy and reproducibility of the results.
CMC
10 \(\mu\)L of fluorescent probe BODIPY (500 \(\mu\)M) dissolved in methanol was combined with varying amounts of P3 in methanol. The resulting mixtures were subjected to solvent evaporation under reduced pressure to remove methanol. Subsequently, PBS was added to the dried residues and sonicated for 5 min to create nanoparticle dispersions. The fluorescent intensity of these dispersions was then measured using a ThermoFisher Varioskan LUX, \(\lambda _{Ex}\) = 475 nm, \(\lambda _{Em}\) = 500 nm. The emission intensities were plotted against the concentrations of P3.
HPLC analysis of compound 3 reacting with SDT
Model compound 3 (0.1 mM) was incubated with sodium dithionite (SDT, 2.5 mM) for 30 min at \(37 ^{\circ }\textrm{C}\) in PBS (pH 7.4) to simulate colonic azoreductase activity. Intact compound 3 (0.1 mM) and 5-ASA (1 mM) were used as standards. Samples were filtered through a 0.22 μm membrane prior to HPLC injection. A volume of 5 \(\mu\)L of each sample was injected into an Agilent 1260 liquid chromatography system equipped with a C18 reverse-phase column (4.6 \(\times\) 75 mm, 5 \(\mu\)m particle size). Compound separation was achieved using a mobile phase gradient of acetonitrile (10–100%) in water at a flow rate of 0.2 mL/min over 7 min. UV detection was performed at 330 nm to quantify the levels of compound 3 and released 5-ASA.
HPLC analysis of 5-ASA release from P3
Gastric contents, SI contents and colonic contents were collected from healthy mice. Following the euthanization of mice with CO2 gas, the gastrointestinal contents were gathered and subsequently mixed with PBS to create a 10% (w/v) suspension. P3 was incubated with intestinal contents for 2, 8, and 12 h and colon contents for different time periods at \(37 ^{\circ }\)C under Ar (3 mg \(\hbox {mL}^{-1}\)P3 in each biofluid). After incubation, samples were centrifuged at a speed of 10,000 rpm for 20 min and then filtered using a 10 kDa ultrafiltration unit to remove polymers and proteins. All collected samples were filtered through a 0.22 \(\mu\)m membrane prior to HPLC injection. The filtrate containing released 5-ASA was injected into an Agilent 1260 HPLC system equipped with a size exclusion chromatographic column (7.8 \(\times\)150 mm, 5 \(\mu\)m particle size). 150 mM PBS (pH 7.0) was used as the mobile phase at a flow rate of 0.35 mL \(\hbox {min}^{-1}\). A sample injection volume of 20 \(\mu\)L was used. 5-ASA was detected using a UV detector at 330 nm based on comparison with retention time of an authentic standard. The released 5-ASA was quantified by integration of peak area.
HPLC analysis of 5-ASA release from SSZ
Gastric contents, SI contents and colonic contents were collected from healthy mice following the same procedure as described above. SSZ was incubated with intestinal contents for 2, 4, 8, and 12 h and colon contents for different time periods at \(37 ^{\circ }\)C under Ar (0.2 mM SSZ in each biofluid). After incubation, samples were centrifuged at a speed of 10,000 rpm for 20 min and then filtered using a 10 kDa ultrafiltration unit to remove proteins. All collected samples were filtered through a 0.22 \(\mu\)m membrane prior to HPLC injection. The filtrate containing released 5-ASA was injected into an Agilent 1260 HPLC system equipped with a size exclusion chromatographic column (7.8 \(\times\)150 mm, 5 \(\mu\)m particle size). MeOH: PBS (1:1, v: v) was used as the mobile phase at a flow rate of 0.35 mL min-1. A sample injection volume of 20 \(\mu\)L was used. 5-ASA was detected using a UV detector at 330 nm based on comparison with retention time of an authentic standard. The released 5-ASA was quantified by integration of peak area.
MS analysis of 5-ASA release from P3
P3 (0.05 mg \(\hbox {mL}^{-1}\)) was incubated in simulated gastric fluid (SGF, pH 2.0), simulated intestinal fluid (SIF, pH 6.8), or SIF containing sodium dithionite (SDT, 0.84 mM) at \(37 ^{\circ }\)C for 0.5 h. Controls included native P3 (0.05 mg \(\hbox {mL}^{-1}\)) and 5-ASA standard (1 mM). After incubation, samples were filtered using a 3 kDa centrifugal filter unit to remove polymers. The filtrate was injected into an Agilent 6520 Q-TOF LC-MS system equipped with an electrospray ionization (ESI) source operating in negative ion mode. Mass spectra were acquired from m/z 150–500. Source parameters were: gas temperature \(300 ^{\circ }\)C, drying gas 9 L \(\hbox {min}^{-1}\), nebulizer 45 psi, capillary voltage 3500 V.
Cell cytotoxic assays
The cell viability was evaluated using the standard Cell Counting Kit-8 (CCK-8) method. Caco-2 or RAW 264.7 cells (\(5 \times 10^4 \hbox{ cells mL}^{-1}\)) were inoculated into Petri dishes with DMEM, 10% fetal bovine serum, 1% penicillin, and 1% streptomycin and cultured in 96-well plates for 24 h, and then different concentrations (0, 0.15, 0.3, 0.6, 1.25, 2.5 and 5 mg \(\hbox {mL}^{-1}\)) of the polymers were introduced and further incubated with the cells for 24 h at \(37 ^{\circ }\)C. Cells were then washed with PBS buffer thrice before being treated with CCK-8 solutions (100 \(\mu\)L, 10%) for 2 h at \(37 ^{\circ }\)C. Finally, the cell viability was obtained by measuring the absorbance at 450 nm.
DSS induced colitis mouse model
In vivo studies were performed on male C57BL/6J mice (7 weeks old, 22–24 g), provided by Zhejiang Vital River Experimental Animal Technology Co. LTD). Mice received the housing process based on normal condition and were given adlibitum distilled water and food. The UC mice model was established by administering 2.5% (w/v) dextran sulfate sodium (DSS, 36–50 kDa) in drinking water for 7 days and induced colonic inflammation. The appearance of weight loss, loose stool, diarrhea, bloody stool or fecal occult blood, and ulcers were regarded as signs of successful modeling.
Retention of NBP@P3 and NB in vivo
The retention time of NBP@P3 and NB was assessed using whole-body fluorescence imaging experiment. The procedure involved the oral administration of NBP@P3 (30 mg \(\hbox {mL}^{-1}\), with 150 \(\mu\)M NBP, 200 \(\mu\)L) and NB (0.5 \(\mu\)mol \(\hbox {kg}^{-1}\), 200 \(\mu\)L) to both healthy and DSS-induced colitic mice. The imaging study was performed at specific time points, including 0.5, 3, 6, 12, and 24 h post-administration. Each mouse group received the respective compound, and the imaging sessions were conducted using an in vivo imaging system equipped with \(\lambda _{Ex}\) = 580 nm, \(\lambda _{Em}\) = 620 nm for NBP and \(\lambda _{Ex}\) = 620 nm, \(\lambda _{Em}\) = 670 nm for Nile Blue fluorescence detection. At desired time, colon and major organs of mice were collected and imaged under the same conditions.
Treatment of the UC mice
The mice were separated into six groups, namely, healthy control group, DSS model group, 5-ASA-treated DSS group, P2-treated DSS group, 5-ASA and P2-treated DSS group and P3-treated DSS group. Control healthy mice received normal drinking water only. The colitic mice received daily oral gavage for 7 days with 200 \(\mu\)L solutions of PBS, 5-ASA (30 mg \(\hbox {kg}^{-1}\)), P2 (270 mg\(\hbox {kg}^{-1}\)), P2 + 5-ASA (270 mg \(\hbox {kg}^{-1}\) + 30 mg \(\hbox {kg}^{-1}\)), or an equivalent 5-ASA dose of P3 (300 mg \(\hbox {kg}^{-1}\)). Mice were monitored daily for changes in body weight, stool consistency, and fecal occult blood as indicators of colitis severity. Mice were euthanized on day 14 for sample collection. At the end of treatment, colon, major organs and feces of mice were collected. All animal studies were conducted under protocols approved by the Institutional Animal Care and Use Committee at Wenzhou Institute, University of Chinese Academy of Sciences.
Weight loss and Disease Activity Index (DAI)
Body weight loss and DAI were monitored daily as indicators of colitis progression. Body weight was measured daily and percent change from day 0 was calculated. Stool consistency and fecal occult blood were evaluated using the following scoring system: Occult blood - 0, no blood; 1, microscopic bleeding; 2, mild bleeding; 3, obvious bleeding; 4, gross bleeding. Stool consistency - 0, normal; 1, loose stool; 2, mild diarrhea; 3, diarrhea; 4, severe diarrhea. DAI was calculated by combining the occult blood score, stool consistency score, and percent body weight change score, which was assigned as follows: 0, none; 1, 1–5% loss; 2, 5–10% loss; 3, 10–20% loss; 4, >20% loss. Disease activity index (DAI) was obtained based on the summation of stool consistency state (0–4), fecal bleeding (0–4), and body weight loss (0–4).
Spleen index
Spleens were harvested immediately after euthanasia. Wet weight of each spleen was measured, and the spleen index was calculated as: spleen weight (mg) / body weight (10 g). Higher spleen index indicates greater systemic inflammation.
Cytokine analysis
On day 14, whole blood was collected via eye bleed under isoflurane anesthesia. Blood was allowed to clot at room temperature for 30 min before centrifuging at 2500 rpm for 20 min at \(4 ^{\circ }\)C to isolate serum. Inflammatory cytokines including interleukin (IL)-1\(\beta\), IL-6, and tumor necrosis factor (TNF)-\(\alpha\) were quantified in serum using mouse-specific ELISA kits per the manufacturer’s protocol.
Histological analysis
After euthanasia, the colon from cecum to anus was removed and length measured. A 0.5 cm portion of distal colon was fixed in 4% paraformaldehyde for 24 h followed by paraffin embedding and sectioning into 4 μm slices. Hematoxylin and eosin (H&E) staining was performed to assess tissue morphology. The images were taken using an upright microscope. Tight junction proteins ZO-1, Claudin-1, and Occludin were imaged by immunofluorescence staining. The images were taken using fluorescent microscope and Confocal Laser Scanning Microscopy (CLSM).
Biocompatibility
For biocompatibility assessment, heart, liver, spleen, lung and kidney were harvested and fixed in 4% paraformaldehyde. Sections were paraffin-embedded, sliced to 4 μm, and H &E stained to evaluate tissue morphology and identify any signs of toxicity.
Conclusion
In summary, this study represents a significant advancement in the realm of UC therapy through the development of a novel drug delivery system. Our research focuses on the rational design of amphiphilic polymeric prodrugs, specifically tailored to optimize 5-ASA release kinetics for colitis therapy. These prodrugs self-assemble into small micellar nanoparticles, offering several advantages: (1) The incorporation of enzyme-cleavable 5-ASA prodrug units in the micelle corona enables specific drug release at the inflamed colonic site while avoiding systemic exposure. (2) The formation of micelles as well as negatively charged surface promotes the enhancing adhesion to diseased mucosa. (3) Additionally, the architecture of the polymeric design allows for controlled modulation of release kinetics.
Both in vitro and in vivo experiments provide compelling evidence of the efficacy of this drug delivery system. In vitro studies demonstrated selective 5-ASA release in the presence of azoreductase activity and validated stable circulation with negligible premature release followed by sustained colonic delivery, in stark contrast to burst release from sulfasalazine prodrugs. Furthermore, in vivo studies using a DSS-induced mouse model of colitis demonstrated that our negatively charged P3 formulation, with a reduced dosage of 5-ASA at just 30 mg \(\hbox {kg}^{-1}\), 60% less than those used in the most advanced research cases, [27, 28, 72] successfully prolonged retention time and surpassed the effectiveness of standard 5-ASA treatments. Instead of compromising the therapeutic efficacy caused by lower dosage, this approach led to a halt in weight loss and even weight gain in treated mice. DAI results further supported the enhanced therapeutic efficacy of P3, as it exhibited earlier recovery and lower DAI scores compared to other treatment groups. Histological analysis showed that P3 preserved colonic morphology and suppressed inflammation effectively, further validating its superior therapeutic potential. While our in vitro and ex vivo data strongly support colon-specific 5-ASA release, we acknowledge the limitation of not directly visualizing drug release in vivo. Future studies employing advanced imaging techniques, such as activatable fluorescent probes linked via azo bonds, could provide real-time visualization of 5-ASA release in the colonic microenvironment.
In conclusion, our polymeric prodrug approach offers a promising avenue for enhanced UC therapy. By addressing the limitations of conventional treatments, such as poor bioavailability, rapid clearance and undesired release profile, our drug delivery system has the potential to significantly improve patient outcomes and quality of life.
References
Ouyang J, Zhang Z, Deng B, Liu J, Wang L, Liu H, Koo S, Chen S, Li Y, Yaremenko AV, et al. Oral drug delivery platforms for biomedical applications. Materials Today; 2023.
Baryakova TH, Pogostin BH, Langer R, McHugh KJ. Overcoming barriers to patient adherence: the case for developing innovative drug delivery systems. Nat Rev Drug Disc. 2023;22(5):387–409.
Wu B, Kulkarni K, Basu S, Zhang S, Hu M. First-pass metabolism via udp-glucuronosyltransferase: a barrier to oral bioavailability of phenolics. J Pharm Sci. 2011;100(9):3655–81.
Date AA, Hanes J, Ensign LM. Nanoparticles for oral delivery: design, evaluation and state-of-the-art. J Control Release. 2016;240:504–26.
Deng B, Liu S, Wang Y, Ali B, Kong N, Xie T, Koo S, Ouyang J, Tao W. Oral nanomedicine: challenges and opportunities. Adv Mater. 2023;1:2306081.
Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Disc. 2018;17(8):559–87.
Clas S-D, Sanchez RI, Nofsinger R. Chemistry-enabled drug delivery (prodrugs): recent progress and challenges. Drug Disc Today. 2014;19(1):79–87.
Matsuoka K, Igarashi A, Sato N, Isono Y, Gouda M, Iwasaki K, Shoji A, Hisamatsu T. Trends in corticosteroid prescriptions for ulcerative colitis and factors associated with long-term corticosteroid use: analysis using japanese claims data from 2006 to 2016. J Crohn’s Colitis. 2021;15(3):358–66.
Salice M, Rizzello F, Calabrese C, Calandrini L, Gionchetti P. A current overview of corticosteroid use in active ulcerative colitis. Exp Rev Gastroenterol Hepatol. 2019;13(6):557–61.
Mikami Y, Tsunoda J, Suzuki S, Mizushima I, Kiyohara H, Kanai T. Significance of 5-aminosalicylic acid intolerance in the clinical management of ulcerative colitis. Digestion. 2023;104(1):58–65.
Louis E, Paridaens K, Al Awadhi S, Begun J, Cheon JH, Dignass AU, Magro F, Márquez JR, Moschen AR, Narula N, et al. Modelling the benefits of an optimised treatment strategy for 5-asa in mild-to-moderate ulcerative colitis. BMJ Open Gastroenterol. 2022;9(1): 000853.
Sehgal P, Colombel J-F, Aboubakr A, Narula N. Systematic review: safety of mesalazine in ulcerative colitis. Aliment Pharmacol Therap. 2018;47(12):1597–609.
Lichtenstein G, Kamm M. 5-aminosalicylate formulations for the treatment of ulcerative colitis-methods of comparing release rates and delivery of 5-aminosalicylate to the colonic mucosa. Aliment Pharmacol Therap. 2008;28(6):663–73.
Raine T, Bonovas S, Burisch J, Kucharzik T, Adamina M, Annese V, Bachmann O, Bettenworth D, Chaparro M, Czuber-Dochan W, et al. Ecco guidelines on therapeutics in ulcerative colitis: medical treatment. J Crohn’s Colitis. 2022;16(1):2–17.
Sands BE, Sandborn WJ, Panaccione R, O’Brien CD, Zhang H, Johanns J, Adedokun OJ, Li K, Peyrin-Biroulet L, Van Assche G, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201–14.
Taku K, Britta S, Chen WS, Ferrante M, Shen B, Bernstein CN, Silvio D, Laurent P-B, Toshifumi H, et al. Ulcerative colitis (primer). Nat Rev. 2020;6:1.
Chaparro M, Gisbert JP. Maintenance therapy options for ulcerative colitis. Exp Opin Pharmacother. 2016;17(10):1339–49.
Panés J, Alfaro I. New treatment strategies for ulcerative colitis. Exp Rev Clin Immunol. 2017;13(10):963–73.
Hu S, Zhao R, Xu Y, Gu Z, Zhu B, Hu J. Orally-administered nanomedicine systems targeting colon inflammation for the treatment of inflammatory bowel disease: latest advances. Journal of Materials Chemistry B. 2024.
Wang R, Cao S, Bashir MEH, Hesser LA, Su Y, Hong SMC, Thompson A, Culleen E, Sabados M, Dylla NP, et al. Treatment of peanut allergy and colitis in mice via the intestinal release of butyrate from polymeric micelles. Nat Biomed Eng. 2023;7(1):38–55.
Fardafshari F, Taymouri S, Minaiyan M, Tavakoli N. Preparing simvastatin nanoparticles by a combination of ph-sensitive and timed-release approaches for the potential treatment of ulcerative colitis. J Biomater Appl. 2022;37(5):859–71.
Yan X, Yang C, Yang M, Ma Y, Zhang Y, Zhang Y, Liu C, Xu Q, Tu K, Zhang M. All-in-one theranostic nano-platform based on polymer nanoparticles for bret/fret-initiated bioluminescence imaging and synergistically anti-inflammatory therapy for ulcerative colitis. J Nanobiotechnol. 2022;20(1):99.
Yang C, Sung J, Long D, Alghoul Z, Merlin D. Prevention of ulcerative colitis by autologous metabolite transfer from colitogenic microbiota treated with lipid nanoparticles encapsulating an anti-inflammatory drug candidate. Pharmaceutics. 2022;14(6):1233.
Geraili A, Xing M, Mequanint K. Design and fabrication of drug-delivery systems toward adjustable release profiles for personalized treatment. View. 2021;2(5):20200126.
Ghezzi M, Pescina S, Padula C, Santi P, Del Favero E, Cantù L, Nicoli S. Polymeric micelles in drug delivery: An insight of the techniques for their characterization and assessment in biorelevant conditions. J Control Release. 2021;332:312–36.
Zhang Y, Wu Y, Yan Y, Ma Y, Tu L, Shao J, Tang X, Chen L, Liang G, Yin L. Dual-targeted nanoparticle-in-microparticle system for ulcerative colitis therapy. Adv Healthcare Mater. 2023;12(31):2301518.
Zhang K, Zhu L, Zhong Y, Xu L, Lang C, Chen J, Yan F, Li J, Qiu J, Chen Y, et al. Prodrug integrated envelope on probiotics to enhance target therapy for ulcerative colitis. Adv Sci. 2023;10(4):2205422.
Peng P, Feng T, Yang X, Nie C, Yu L, Ding R, Zhou Q, Jiang X, Li P. Gastrointestinal microenvironment responsive nanoencapsulation of probiotics and drugs for synergistic therapy of intestinal diseases. ACS Nano. 2023;17(15):14718–30.
Huckaby JT, Lai SK. Pegylation for enhancing nanoparticle diffusion in mucus. Adv Drug Deliv Rev. 2018;124:125–39.
Maisel K, Ensign L, Reddy M, Cone R, Hanes J. Effect of surface chemistry on nanoparticle interaction with gastrointestinal mucus and distribution in the gastrointestinal tract following oral and rectal administration in the mouse. J Control Rel. 2015;197:48–57.
Subramanian DA, Langer R, Traverso G. Mucus interaction to improve gastrointestinal retention and pharmacokinetics of orally administered nano-drug delivery systems. J Nanobiotechnol. 2022;20(1):1–23.
Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–63.
Bohrey S, Chourasiya V, Pandey A. Polymeric nanoparticles containing diazepam: preparation, optimization, characterization, in-vitro drug release and release kinetic study. Nano Convergence. 2016;3(1):1–7.
Son G-H, Lee B-J, Cho C-W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J Pharmac Invest. 2017;47:287–96.
Tian B, Liu S, Lu W, Jin L, Li Q, Shi Y, Li C, Wang Z, Du Y. Construction of ph-responsive and up-conversion luminescent nayf4: Yb3+/er3+@ sio2@ pmaa nanocomposite for colon targeted drug delivery. Sci Rep. 2016;6(1):21335.
Tang H, Xiang D, Wang F, Mao J, Tan X, Wang Y. 5-asa-loaded sio2 nanoparticles-a novel drug delivery system targeting therapy on ulcerative colitis in mice. Mol Med Rep. 2017;15(3):1117–22.
Wang N, Shao L, Lu W, Fang W, Zhang Q, Sun L, Gao S, Zhu Q, Chen S, Hu R. 5-aminosalicylic acid ph sensitive core-shell nanoparticles targeting ulcerative colitis. J Drug Del Sci Technol. 2022;74: 103578.
Tang S, Yang R, Gao Y, Zhu L, Zheng S, Zan X. Hydrazone-based amphiphilic brush polymer for fast endocytosis and ros-active drug release. ACS Macro Lett. 2023;12(5):639–45.
Naren G, Hsu C-W, Li S, Morimoto M, Tang S, Hernando J, Guirado G, Irie M, Raymo FM, Sundén H, et al. An all-photonic full color rgb system based on molecular photoswitches. Nat Commun. 2019;10(1):3996.
Tang S, Zhang Y, Dhakal P, Ravelo L, Anderson CL, Collins KM, Raymo FM. Photochemical barcodes. J Am Chem Soc. 2018;140(13):4485–8.
Tang S, Zhang Y, Thapaliya ER, Brown AS, Wilson JN, Raymo FM. Highlighting cancer cells with halochromic switches. ACS Sens. 2017;2(1):92–101.
Pietropaolo A, Tang S, Raymo FM. Free-energy predictions and absorption spectra calculations for supramolecular nanocarriers and their photoactive cargo. Nanoscale. 2017;9(15):4989–94.
Tang S, Donaphon B, Levitus M, Raymo FM. Structural implications on the properties of self-assembling supramolecular hosts for fluorescent guests. Langmuir. 2016;32(34):8676–87.
Zhang Y, Swaminathan S, Tang S, Garcia-Amorós J, Boulina M, Captain B, Baker JD, Raymo FM. Photoactivatable bodipys designed to monitor the dynamics of supramolecular nanocarriers. J Am Chem Soc. 2015;137(14):4709–19.
Swaminathan S, Fowley C, Thapaliya ER, McCaughan B, Tang S, Fraix A, Captain B, Sortino S, Callan JF, Raymo FM. Supramolecular nanoreactors for intracellular singlet-oxygen sensitization. Nanoscale. 2015;7(33):14071–9.
Tang S, Gao Y, Wang W, Wang Y, Liu P, Shou Z, Yang R, Jin C, Zan X, Wang C, et al. Self-report amphiphilic polymer-based drug delivery system with ros-triggered drug release for osteoarthritis therapy. ACS Macro Lett. 2023;13(1):58–64.
Davoudi Z, Peroutka-Bigus N, Bellaire B, Wannemuehler M, Barrett TA, Narasimhan B, Wang Q. Intestinal organoids containing poly (lactic-co-glycolic acid) nanoparticles for the treatment of inflammatory bowel diseases. J Biomed Mater Res, Part A. 2018;106(4):876–86.
Zhao J, Zhang B, Mao Q, Ping K, Zhang P, Lin F, Liu D, Feng Y, Sun M, Zhang Y, et al. Discovery of a colon-targeted azo prodrug of tofacitinib through the establishment of colon-specific delivery systems constructed by 5-asa-paba-mac and 5-asa-paba-diamine for the treatment of ulcerative colitis. J Med Chem. 2022;65(6):4926–48.
Carceller E, Salas J, Merlos M, Giral M, Ferrando R, Escamilla I, Ramis J, García-Rafanell J, Forn J. Novel azo derivatives as prodrugs of 5-aminosalicylic acid and amino derivatives with potent platelet activating factor antagonist activity. J Med Chem. 2001;44(18):3001–13.
Kim W, Nam J, Lee S, Jeong S, Jung Y. 5-aminosalicylic acid azo-linked to procainamide acts as an anticolitic mutual prodrug via additive inhibition of nuclear factor kappab. Mol Pharm. 2016;13(6):2126–35.
Jeong S, Lee H, Kim S, Ju S, Kim W, Cho H, Kim HY, Heo G, Im E, Yoo J-W, et al. 5-aminosalicylic acid azo-coupled with a gpr109a agonist is a colon-targeted anticolitic codrug with a reduced risk of skin toxicity. Mol Pharm. 2019;17(1):167–79.
Dutta P, Mukherjee K, Saha A, Das A, Badwaik HR, Giri TK. Colonic delivery of surface charge decorated nanocarrier for ibd therapy. J Drug Del Sci Technol. 2022;76: 103754.
Guo L, Zhuge Y, Yang L, Qiu H, Liu J, Wang P. A two-photon mitochondria-targeting azo reductase probe for imaging in tumor cells and mice. Dyes Pigm. 2023;218: 111512.
Li J-J, Rong R-X, Yang Y, Hu Z-Y, Hu B, Zhao Y-Y, Li H-B, Hu X-Y, Wang K-R, Guo D-S. Triple targeting host-guest drug delivery system based on lactose-modified azocalix [4] arene for tumor ablation. Mater Horizons. 2023;10(5):1689–96.
Hou X, Chang Y-X, Yue Y-X, Wang Z-H, Ding F, Li Z-H, Li H-B, Xu Y, Kong X, Huang F, et al. Supramolecular radiosensitizer based on hypoxia-responsive macrocycle. Adv Sci. 2022;9(6):2104349.
Sangekar S, Vadino W, Chaudry I, Parr A, Beihn R, Digenis G. Evaluation of the effect of food and specific gravity of tablets on gastric retention time. Int J Pharm. 1987;35(3):187–91.
Kedzierewicz F, Thouvenot P, Lemut J, Etienne A, Hoffman M, Maincent P. Evaluation of peroral silicone dosage forms in humans by gamma-scintigraphy. J Control Rel. 1999;58(2):195–205.
Intawongse M, Dean JR. In-vitro testing for assessing oral bioaccessibility of trace metals in soil and food samples. TrAC, Trends Anal Chem. 2006;25(9):876–86.
Sunesen VH, Vedelsdal R, Kristensen HG, Christrup L, Müllertz A. Effect of liquid volume and food intake on the absolute bioavailability of danazol, a poorly soluble drug. Eur J Pharm Sci. 2005;24(4):297–303.
Roowi S, Stalmach A, Mullen W, Lean ME, Edwards CA, Crozier A. Green tea flavan-3-ols: colonic degradation and urinary excretion of catabolites by humans. J Agric Food Chem. 2010;58(2):1296–304.
Koyama E, Kitazawa K, Ohori Y, Izawa O, Kakegawa K, Fujino A, Ui M. In vitro metabolism of the glycosidic sweeteners, stevia mixture and enzymatically modified stevia in human intestinal microflora. Food Chem Toxicol. 2003;41(3):359–74.
Cha KH, Koo SY, Lee D-U. Antiproliferative effects of carotenoids extracted from chlorella ellipsoidea and chlorella vulgaris on human colon cancer cells. J Agric Food Chem. 2008;56(22):10521–6.
Chevalier A, Renard P-Y, Romieu A. Azo-based fluorogenic probes for biosensing and bioimaging: Recent advances and upcoming challenges. Chemistry. 2017;12(16):2008–28.
Yanai H, Hoshikawa S, Moriiwa Y, Shoji A, Yanagida A, Matsumoto T. A fluorinated carbanionic substituent for improving water solubility and lipophilicity of fluorescent dyes. Angew Chem. 2021;133(10):5228–32.
Yasukawa K, Hirago A, Yamada K, Tun X, Ohkuma K, Utsumi H. In vivo redox imaging of dextran sodium sulfate-induced colitis in mice using overhauser-enhanced magnetic resonance imaging. Free Radical Biol Med. 2019;136:1–11.
Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (dss)-induced colitis in mice. Curr Prot Immunol. 2014;104(1):15–25.
Hirai F, Matsui T, Aoyagi K, Inoue N, Hibi T, Oshitani N, Fujii H, Kobayashi K, Suzuki Y, Tanaka S. Validity of activity indices in ulcerative colitis: comparison of clinical and endoscopic indices. Dig Endosc. 2010;22(1):39–44.
Veenstra JP, Vemu B, Tocmo R, Nauman MC, Johnson JJ. Pharmacokinetic analysis of carnosic acid and carnosol in standardized rosemary extract and the effect on the disease activity index of dss-induced colitis. Nutrients. 2021;13(3):773.
Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating intestinal inflammation in dss-induced model of ibd. J Visual Exp. 2012;60:3678.
Kim Y, Lim HJ, Jang H-J, Lee S, Jung K, Lee SW, Lee S-J, Rho M-C. Portulaca oleracea extracts and their active compounds ameliorate inflammatory bowel diseases in vitro and in vivo by modulating tnf-\(\alpha\), il-6 and il-1\(\beta\) signalling. Food Res Int. 2018;106:335–43.
Szkaradkiewicz A, Marciniak R, Chudzicka-Strugała I, Wasilewska A, Drews M, Majewski P, Karpiński T, Zwoździak B. Proinflammatory cytokines and il-10 in inflammatory bowel disease and colorectal cancer patients. Archivum immunologiae et therapiae experimentalis. 2009;57:291–4.
Zhao P, Xia X, Xu X, Leung KKC, Rai A, Deng Y, Yang B, Lai H, Peng X, Shi P, et al. Nanoparticle-assembled bioadhesive coacervate coating with prolonged gastrointestinal retention for inflammatory bowel disease therapy. Nat Commun. 2021;12(1):7162.
Acknowledgements
The authors would like to thank Rongqi Xia from Shiyanjia Lab (www.shiyanjia.com) for the HRMS analysis. Figs. 1a and 3a were created using materials sourced from Figdraw (https://www.figdraw.com).
Funding
This work was financially supported by startup funding from the Wenzhou Institute of UCAS (WIUCASQD2021046), Wenzhou Science and Technology Project (2023Y0847) and Zhejiang Provincial Natural Science Foundation of China (LQ24H120002).
Author information
Authors and Affiliations
Contributions
Sicheng Tang and Wenchao Wang contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments conducted in this study were performed in strict accordance with the guidelines and regulations set forth by the Wenzhou Institute, UCAS. This study was approved by the WIUCAS Animal Ethics Committee, with the approval certificate number WIUCAS23033101.
Competing interests
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, S., Wang, W., Wang, Y. et al. Sustained release of 5-aminosalicylic acid from azoreductase-responsive polymeric prodrugs for prolonged colon-targeted colitis therapy. J Nanobiotechnol 22, 468 (2024). https://doi.org/10.1186/s12951-024-02724-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-024-02724-w