
Abstract
Methods
Single-cell transcriptomics and high-throughput transcriptomics were used to screen factors significantly correlated with intervertebral disc degeneration (IDD). Expression changes of CFIm25 were determined via RT-qPCR and Western blot. NP cells were isolated from mouse intervertebral discs and induced to degrade with TNF-α and IL-1β. CFIm25 was knocked out using CRISPR-Cas9, and CFIm25 knockout and overexpressing nucleus pulposus (NP) cell lines were generated through lentiviral transfection. Proteoglycan expression, protein expression, inflammatory factor expression, cell viability, proliferation, migration, gene expression, and protein expression were analyzed using various assays (alcian blue staining, immunofluorescence, ELISA, CCK-8, EDU labeling, transwell migration, scratch assay, RT-qPCR, Western blot). The GelMA-HAMA hydrogel loaded with APET×2 polypeptide and sgRNA was designed, and its effects on NP regeneration were assessed through in vitro and mouse model experiments. The progression of IDD in mice was evaluated using X-ray, H&E staining, and Safranin O-Fast Green staining. Immunohistochemistry was performed to determine protein expression in NP tissue. Proteomic analysis combined with in vitro and in vivo experiments was conducted to elucidate the mechanisms of hydrogel action.
Results
CFIm25 was upregulated in IDD NP tissue and significantly correlated with disease progression. Inhibition of CFIm25 improved NP cell degeneration, enhanced cell proliferation, and migration. The hydrogel effectively knocked down CFIm25 expression, improved NP cell degeneration, promoted cell proliferation and migration, and mitigated IDD progression in a mouse model. The hydrogel inhibited inflammatory factor expression (IL-6, iNOS, IL-1β, TNF-α) by targeting the p38/NF-κB signaling pathway, increased collagen COLII and proteoglycan Aggrecan expression, and suppressed NP degeneration-related factors (COX-2, MMP-3).
Conclusion
The study highlighted the crucial role of CFIm25 in IDD and introduced a promising therapeutic strategy using a porous spherical GelMA-HAMA hydrogel loaded with APET×2 polypeptide and sgRNA. This innovative approach offers new possibilities for treating degenerated intervertebral discs.
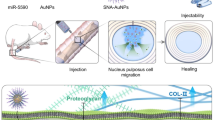
Graphical Abstract

Molecular mechanism of GCA targeting CFIm25 in NP cells to promote regeneration of degenerated intervertebral disc NP
Similar content being viewed by others


Introduction
Intervertebral disc degeneration (IDD) is a common degenerative disease of the spine that is globally increasing in prevalence [1,2,3]. The intervertebral disc is an important component of the spine that maintains its normal function by absorbing and dispersing spinal load [4]. However, the long-term impact of mechanical, biological, and chemical factors leads to IDD, causing it to lose its normal structure and function [5, 6]. IDD severely affects an individual’s quality of life and health, causing pain, limited mobility, and neurological disorders [7,8,9]. Therefore, exploring the etiology of IDD and finding effective treatment strategies are important directions of research.
The etiology of IDD is extremely complex, involving interactions between multiple cells and molecules [3, 10, 11]. Studies have found significant infiltration of inflammatory cells and release of inflammatory factors during the progression of IDD [12,13,14]. These factors can activate matrix-degrading enzymes, leading to the degradation of intervertebral disc matrix proteins and subsequent structural and functional damage [15, 16]. In addition, IDD also involves various pathological processes such as cell apoptosis, neovascularization, and abnormal expression of nerve growth factors [17,18,19]. Therefore, an in-depth investigation of key factors and molecules in the pathogenesis of IDD is of great significance in the search for effective treatment strategies.
Intervertebral discs (IVDs) consist of the nucleus pulposus (NP), annulus fibrosus, and upper and lower cartilaginous endplates. The NP tissue, a highly hydrated gel-like structure, exhibits characteristics of shock absorption and low-load endurance [20]. IVD degeneration commonly involves a reduction in hydrophilic substances such as type II collagen (COL-II) and aggrecan, upregulation of matrix metalloproteinases (MMPs) in the disc matrix, increased oxidative stress, decreased functional NP cell population, and fibrosis of the NP tissue [21, 22]. Consequently, IVD degeneration is considered a multifaceted pathological process at the molecular, cellular, and tissue levels.
Hydrogels have significant applications in the biomedical field, particularly in tissue engineering and drug delivery systems. Serving as a three-dimensional scaffold, hydrogels provide an environment mimicking the extracellular matrix, facilitating cell growth and tissue regeneration [23]. Biologically active hydrogels are preferred materials for IVD repair due to their excellent biocompatibility and rheological properties [24]. Additionally, hydrogels effectively encapsulate and protect gene editing tools such as the CRISPR-Cas9 system, enhancing stability and efficiency within target cells or tissues [25].
Single-cell RNA sequencing (scRNA-seq) is a high-throughput technique enabling genome and transcriptome sequencing and analysis at a single-cell level [26]. It identifies unique cell subpopulations, elucidates cellular changes, and becomes increasingly vital in fields like cancer biology, developmental biology, and neurology [27,28,29].
Hyaluronic acid-methacryloyl (HAMA) is a linear polysaccharide composed of repeating units of β-1,4-D-glucuronic acid and β-1,3-N-acetyl-D-glucosamine, promoting wound healing, regulating tissue formation, correcting inflammatory microenvironments, but hindering cell adhesion, thus limiting biological behaviors like proliferation, elongation, migration, and differentiation [30]. However, GelMA hydrogels containing a specific arginine-glycine-aspartic acid (RGD) peptide sequence provide cell adhesion sites by binding multiple integrins, overcoming the primary limitations of HAMA [31]. Building a bipolymeric network hydrogel with GelMA and HAMA can address the limitations of single-component hydrogels, benefiting the improvement of degenerative disc injury [32].
In this study, through single-cell RNA sequencing (scRNA-seq) analysis, we identified a significant upregulation of CFIm25 in the NP tissue of IDD. Therefore, we have chosen to investigate CFIm25 as a key factor in this process. To elucidate the mechanism of action of CFIm25 further and develop new therapeutic strategies, we designed and prepared a novel porous spherical GelMA-HAMA hydrogel loaded with APET×2 peptide and sgRNA. Through in vitro and in vivo experiments, we found that this hydrogel effectively downregulates CFIm25 expression, enhances proliferation and migration capabilities of NP cells, thus slowing the progression of IDD. This presents a new avenue for IDD treatment. The aim of our study is to explore the mechanism of promoting NP cell regeneration targeting CFIm25, utilizing a porous spherical GelMA-HAMA hydrogel loaded with APET×2 peptide and sgRNA. If successful clinical therapeutic strategies are developed, it could provide a new choice for the treatment of degenerative disc disease, ultimately improving patients’ quality of life. Moreover, further investigating the etiology of IDD could provide insights into the treatment strategies for other degenerative skeletal diseases, possessing significant scientific and clinical implications.
Therefore, the aim of this study is to explore the mechanism of promoting intervertebral disc NP regeneration by targeting CFIm25 in NP cells using the porous spherical GelMA-HAMA hydrogel loaded with APET×2 polypeptide and sgRNA. Successful development of clinically applicable treatment strategies will provide new options for the treatment of degenerative IVD, improving patients’ quality of life. Furthermore, in-depth research into the etiology of IDD can shed light on the treatment strategies for other degenerative skeletal diseases, giving it significant scientific and clinical implications.
Materials and methods
Animal ethic statement
This study strictly adheres to internationally recognized animal welfare and ethics guidelines. All experimental protocols have been approved by Honghui Hospital, Xi’an Jiaotong University’s ethical review committee. We have taken great care to ensure the welfare of the mice involved in the experiment. All animals are fed according to appropriate standards and live in suitable environmental conditions. In conducting the experimental procedures, every possible measure has been taken to minimize the pain and discomfort of the animals. At the conclusion of the experiment, humane euthanasia was performed on the animals.
Construction of IDD model in mice
For this study, 6–8 week-old SPF-grade C57BL/6J male mice (219, Beijing Weitonglihua Experimental Animal Technology Co., Ltd., Beijing, China) weighing between 18 and 25 g were used. All mice were housed in individual cages in an SPF-grade animal facility with a controlled lighting schedule of 12 h of darkness and 12 h of light, humidity at 60% ∼ 65%, and temperature at 22 ∼ 25 ℃. Mice had ad libitum access to food and water, and after one week of adaptation, they were observed for their health before the start of the experiment.
The method for constructing the IDD animal model in mice is as follows: Mice were deeply anesthetized using a 2% isoflurane-O2 mixture (1 L/min, R510-22-10, RWD, Shenzhen, China). Under sterile conditions, the experimental mice were placed on a surgical table, and the intervertebral disc area was exposed. The intervertebral disc on one side was punctured using a 27G needle to induce IDD. To ensure the efficacy of the degeneration, the needle was rotated 360° after puncture and held for 30 s. After modeling, the success of the procedure was confirmed through radiological examination and histological analysis. For the control group mice, similar surgical procedures were performed, but IDD was not induced. After completing all surgeries, mice received appropriate postoperative care and monitoring [33, 34].
X-ray imaging examination
The mice were anesthetized with 2% isoflurane (1 L/min) prior to the examination and were positioned in a prone position for the imaging procedure. X-ray images were captured using an exposure time of 40 s, a distance of 40 cm, and a setting of 3 mA/35 kV on Philips X-ray film. The height of the intervertebral space before and after the surgery was measured and expressed as the Intervertebral Disc Height Index (DHI). Surgimap 2.3.2.1 software (Nemaris, New York) was utilized for the measurements. For detailed measurement and calculation methods, refer to Figure S4A [35].
H&E staining
Hematoxylin and eosin (H&E) staining was performed using the Hematoxylin and Eosin Staining Kit (C0105S, Beyotime, Shanghai, China) to observe the pathological changes in the intervertebral disc tissue of mice. Partial intervertebral disc tissue from mice was fixed in 4% paraformaldehyde, followed by dehydration, transparency, and embedding in paraffin. 5 μm thick sections were obtained using a microtome and then subjected to deparaffinization, hydration, staining with hematoxylin, rinsing with distilled water, immersion in 95% ethanol, and subsequent staining with eosin. Differentiation with 70% hydrochloric acid ethanol, dehydration, and transparency was performed before finally mounting the sections with neutral gum. The morphological changes in the intervertebral disc tissue of the mice were observed using an optical microscope [35].
Safranin O-Fast green staining
The Safranin O-Fast Green Staining kit (G1371, Beijing Solabio Technology Co., Ltd., Beijing, China) was used to stain and observe the distribution of acidophilic bone and basophilic cartilage in the intervertebral disc. Following the instructions provided, the specific staining procedure was performed as follows: initially, the slides were stained with Safranin O for one hour; excess dye was then removed by rinsing with distilled water, and subsequently, gradient ethanol (50 − 80%) was used for decolorization. Subsequently, the slides were stained with Fast Green for 60 s and dehydrated using absolute ethanol. Finally, the sections were mounted in a clean xylene-based mounting medium for 5 min, followed by immediate sealing with neutral resin. The staining results were evaluated using a histological scoring method described in the literature, which assessed the staining outcomes from five different perspectives. Each category’s score ranged from 1 to 3, with a cumulative score between 5 and 15, indicating a higher score corresponding to a higher degree of degeneration [36, 37].
Immunohistochemical staining
The paraffin blocks were cooled on ice or in a refrigerated fridge set at 4 °C before embedding and sectioning. The paraffin sections were left to dry overnight while the slides were placed in an oven set at 60 °C and baked for 20 min. Subsequently, the sections were soaked in xylene for 10 min, followed by another 10-minute immersion after changing the xylene. After 5 min of hydration in absolute alcohol, a second hydration was conducted by replacing the absolute alcohol with fresh absolute alcohol for another 5 min. The sections were then sequentially hydrated in 95% and 70% ethanol, with a 10-minute soak in each type of alcohol, and finally rinsed in distilled water for 5 min. The sections were immersed in citrate buffer (pH 6.0) and heated in a high-power microwave oven for 8 min, then cooled to room temperature. The slides were washed thrice in PBS (pH 7.2–7.6), each for 3 min. To inactivate endogenous peroxidase, 3% H2O2 was added and left at room temperature for 10 min. The sections were subsequently washed three times in PBS, each time for 3 min. The sections were blocked with normal goat serum blocking solution (E510009, SBS Genetech, Shanghai, China) at room temperature for 20 min.
After incubation, the sections were incubated overnight at 4 °C with primary antibodies against COLII, Aggrecan, MMP-3, COX-2, and CFIm25 (ab34712, ab216965, ab52915, ab179800, and ab183660, respectively, Abcam, UK), diluted at 1:100, 1:100, 1:1000, 1:100, and 1:500, respectively. The sections were then washed three times with PBS and incubated with secondary antibodies, goat anti-rabbit IgG (ab6721, diluted at 1:5000, Abcam, Cambridge, UK), for 30 min. Afterward, the sections were incubated with SABC (Streptavidin-Biotin Complex, P0603, Beyotime, Shanghai, China) at 37 °C for 30 min. DAB chromogen (P0203, Beyotime, Shanghai, China) was added to each specimen, followed by a 6-minute incubation for color development. Counterstaining was performed with a hematoxylin solution for 30 s. The sections were subsequently dehydrated in 70%, 80%, 90%, 95% ethanol, and finally in absolute ethanol, each for 2 min. The sections were then immersed in xylene twice for 5 min before being mounted with neutral resin. Observations and statistical analysis were conducted using a BX63 upright microscope (Olympus, Japan), with five random high-power fields selected per slide. Image-Pro Plus 6.0 software was utilized to analyze the average pixel density [36]. This experiment was repeated three times.
Preparation of IVD tissue for NP
Lumbar paravertebral exposure surgery was performed on male C57BL/6J mice (n = 4, aged 6–8 weeks). In the experimental group, mice underwent unilateral IVD injury using a 27G needle, while the sham control group underwent the same lumbar paravertebral exposure surgery without injury. At 2, 4, and 8 weeks post-surgery, euthanasia was conducted, and NP tissue was isolated and pooled from the intervertebral disc, ensuring the removal of peripheral muscles, ligaments, and attachments (Figure S1). The groups included the Sham group (n = 1, sham surgery), the 2-week recovery group (n = 1, 2 weeks post-surgery), the 4-week recovery group (n = 1, 4 weeks post-surgery), and the 8-week recovery group (n = 1, 8 weeks post-surgery). The isolated NP tissue was digested in a culture medium containing 0.2% type II collagenase (17101015, Thermo Fisher, USA) and 0.3% trypsin (10165921001, Sigma-Aldrich, UK), followed by centrifugation at 1000 rpm for 10 min at 37℃ in a 5% CO2 environment to obtain cell pellets. The culture medium was removed, and cells were resuspended in PBS. The suspension was filtered through a 70 μm filter to remove unwanted cell fragments. After another centrifugation at 1000 rpm for 10 min, the resulting cell pellet was resuspended in PBS, obtaining single-cell transcriptome sequencing (scRNA-seq) samples [33].
scRNA-seq
We collected samples of mouse NP tissue at 2 weeks, 4 weeks, and 8 weeks after model simulation. The Sham group represents normal mouse NP tissue, while the IDD group represents IDD in mouse NP tissue. Each group consisted of one sample. The cell viability and integrity of the samples were assessed and counted using a microscope and flow cytometer. Qualified individual cells were captured using the C1 Single-Cell Auto Prep System (Fluidigm, Inc., South San Francisco, CA, USA) (Figure S1). After cell capture, mRNA was extracted, and cDNA was synthesized through reverse transcription. The cDNA, post-lysis, and reverse transcription were pre-amplified on a microfluidic chip for subsequent sequencing. The amplified cDNA was used to construct libraries and subjected to single-cell sequencing on the HiSeq 4000 Illumina platform, with parameters set as paired-end reads, read length of 2 × 75 bp, and approximately 20,000 reads per cell [38].
scRNA-seq data analysis
The data was analyzed using the “Seurat” package in R software. Quality control criteria were set as 200 < nFeature_RNA < 5000 and percent.mt < 20 to filter out highly variable genes. To reduce the dimensionality of the scRNA-Seq dataset, a Principal Component Analysis (PCA) was performed on the top 2000 highly variable genes. The first 20 principal components were selected for downstream analysis using the Elbowplot function in the Seurat package. The main cell subgroups were identified using the FindClusters function in Seurat with the default resolution set at res = 1. The UMAP algorithm was then applied to achieve nonlinear dimensionality reduction of the scRNA-seq sequencing data. The Seurat package was utilized to identify marker genes for various cell subgroups, and the “SingleR” package was used for cell annotation. Cell communication analysis was conducted using the “CellChat” package in R, while cell trajectory analysis was performed with the “Monocle2” package. Differential expression genes (DEGs) in the scRNA-Seq dataset were identified using the “Limma” package in R, focusing on DEGs between different samples [39, 40].
Preparation of IVD tissue for high-throughput transcriptome sequencing
Male C57BL/6J mice (n = 3), aged 6–8 weeks, underwent surgical exposure of the L4-L6 lumbar vertebrae through the peritoneum. Single-sided injury to the L4-5 and L5-6 IVDs was performed using a 27G needle. After a 4-week recovery period, the mice were euthanized, and NP tissue was isolated and pooled from either L4-5 and L5-6 discs (IDD group, representing IDD) or L2-3 and L3-4 discs (Control group, Normal control). Peripheral muscles, ligaments, and attachments were carefully removed during tissue dissection (Figure S2). The isolated NP tissue was digested in a culture medium containing 0.2% type II collagenase and 0.3% trypsin at 37 °C and 5% CO2 for 4 h. The culture medium was then centrifuged at 1000 rpm for 10 min to obtain a cell pellet. The culture medium was aspirated, and the cells were resuspended in PBS. The suspension was filtered through a 70 μm filter to remove unwanted cell debris. Subsequently, the cell pellet was resuspended in PBS after centrifugation at 1000 rpm for 10 min to obtain the high-throughput transcriptome sequencing samples.
RNA purity and integrity were verified using the Nanodrop ND-1000 spectrophotometer (Thermo Fisher) to measure the RNA sample concentration by OD260/280, ensuring the absence of protein and organic contamination. The RNA concentration was determined using the Qubit RNA Assay Kit (Q33221, Thermo Fisher, USA). Total RNA samples meeting the following criteria were used for subsequent experiments: RNA integrity number (RIN) ≥ 7.0 and 28 S:18 S ratio ≥ 1.5 [33, 41].
High-throughput transcriptome sequencing
The sequencing libraries were generated and sequenced by CapitalBio Technology (Beijing, China). A total of 5 µg of RNA was used for each sample. Briefly, we employed the Ribo-Zero Magnetic Kit (MRZG12324, Epicentre, USA) to remove ribosomal RNA (rRNA) from the total RNA. Subsequently, the Illumina NEB Next Ultra RNA Library Prep Kit (E7760S, NEB, USA) was utilized to construct the sequencing libraries. The RNA was fragmented using NEB Next First Strand Synthesis Reaction Buffer (5x), resulting in fragment lengths of approximately 300 base pairs (bp). Next, the first-strand cDNA was synthesized using reverse transcription primers and random primers, followed by the preparation of the second-strand cDNA in the second-strand synthesis reaction buffer containing dUTP Mix (10x). The cDNA fragments were subjected to end repair, including the addition of polyA tails and ligating sequencing adapters. After the ligation of Illumina sequencing adapters, the second strand of the cDNA was digested using the USER enzyme (M5508, NEB, USA) to construct a strand-specific library. Subsequently, the library DNA was amplified and purified, followed by PCR enrichment. Finally, library identification was conducted using the Agilent 2100 system, and quantification was performed using the KAPA Library Quantification Kit (KK3605, Merck, USA). Ultimately, paired-end sequencing was carried out on the Illumina NextSeq CN500 platform [42, 43].
Transcriptome sequencing data analysis
The raw sequencing data was examined for the quality of paired-end reads using FastQC software v0.11.8. Preprocessing of the raw data involved removing Illumina sequencing adapters and poly (A) tails sequences using Cutadapt software 1.18. A perl script was employed to eliminate reads with an N content exceeding 5%. Furthermore, the FASTX Toolkit software 0.0.13 was used to extract reads with a base quality of at least 20, accounting for 70% of the bases. The BBMap software was utilized to correct the paired-end sequences. Eventually, the filtered high-quality read fragments were aligned to the mouse reference genome using hisat2 software (0.7.12).
Differential analysis was performed on mRNA read counts using the “Limma” package in the R programming language. The criteria for selecting differentially expressed genes were set as |log2FC| > 1 and P.value < 0.05. The “heatmap” package in R generated a heat map displaying the intersecting genes. The protein-protein interaction (PPI) relationships of key factors were analyzed using the STRING database (https://string-db.org/), with a minimum required interaction score set to 0.700. The Cytoscape 3.5.1 software was used to visualize the network of interaction relationships, and the built-in CytoHubba tool was employed to identify core genes.
For the aforementioned intersecting genes, functional enrichment analysis of Gene Ontology (GO) was conducted using the “ClusterProfiler” package in R, including analysis of biological processes (BP), molecular functions (MF), and cellular components (CC). The results of GO enrichment were visualized through bubble plots and circle plots, with a screening criterion of P < 0.05. Additionally, based on the p-values, the “ClusterProfiler” package in R was used to conduct the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis for the candidate targets. The results of the KEGG enrichment analysis were illustrated through bubble plots and circle plots [41].
Isolation and handling of mouse NP cells
After euthanizing the mice, the skin, muscles, and other soft tissues from the lumbar and tail regions were aseptically removed. The intervertebral disc tissue was extracted and washed several times with PBS to eliminate blood and impurities. The NP tissue was then dissected and placed in DMEM/F12 medium (11320033, Thermo Fisher, USA). It was transferred to a 15 mL centrifuge tube and centrifuged at 1000 rpm for 5 min to remove the supernatant, retaining the pellet. PBS was added for three additional washes. The tissue was minced and placed in a digestion solution containing 0.25% trypsin (25200056, Thermo Fisher, USA) and EDTA and digested at 37℃ for 30 min. During digestion, pipetting was required to separate cells. After digestion, the supernatant was removed by centrifugation at 1000 rpm for 5 min, followed by three washes with PBS. Subsequently, the tissue was exposed to DMEM/F12 medium containing 0.1% type II collagenase at 37℃ for 4 h, followed by three washes with PBS. Finally, the cells were seeded in appropriate culture plates using DMEM/F12 complete culture medium containing 10% FBS (10100147 C, Thermo Fisher, USA) and 1% antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin, 15140163, Thermo Fisher, USA), and cultured at 37℃ with 5% CO2. The culture medium needed to be regularly replaced, and cell growth was monitored [36, 44].
The isolated NP cells were seeded in a 12-well plate. Once the cells adhered to the surface, they were treated with IL-1β (10 ng/mL, HY-P7073, MedChemExpress, USA) or TNF-α (50 ng/mL, HY-P7090, MedChemExpress, USA) for 48 h to induce NP cell degeneration. After treatment, fresh culture medium was replaced, or cells and culture medium were collected after 24 h for subsequent analysis and experiments, such as cell viability assays and gene and protein expression analysis, to evaluate the effectiveness of the degeneration model [45].
Immunofluorescence staining
In addition to the culture medium, cells were rinsed three times with PBS for 2 min each time. They were then fixed with − 20℃ ice-cold methanol for 30 min, and excess methanol was removed. After that, they were washed three times with PBS for 5 min each time. The cells were treated with 0.1% Triton X-100 at room temperature for 15 min, followed by three washes with PBS for 5 min each time. Blocking was performed with BSA for 30 min, and then rabbit anti-COLII or Aggrecan antibodies (catalog numbers PA5-99159 and PA1-1746, respectively, diluted at 1:500, Thermo Fisher, USA) were added and incubated at 37℃ for 60 min. The cells were washed with PBS for 5 min three times. Goat anti-rabbit secondary antibodies labeled with FITC (A-11008 or A-11012, diluted at 1:500, Abcam, UK) were added and incubated at 37℃ in the dark for 60 min, followed by three washes with PBS for 3 min each time. The cells were stained with DAPI for 10 min, and then excessive DAPI was removed by washing with PBS. Finally, a 20 µL mounting medium was added to let it dry before observing and capturing images with a fluorescence microscope. For quantitative analysis, the fluorescence coverage area in six randomly selected fields of view was measured using a 40× objective lens, and the average was calculated [35].
Alcian blue staining
NP cells were uniformly seeded in a 6-well plate, with each well containing a 2 mL complete culture medium to provide optimal growth conditions. Alcian Blue staining of cells was performed using the alcian blue staining kit (G1563, Beijing Solabel Science & Technology Co., Ltd., Beijing, China), following the detailed instructions provided by the manufacturer. The fluorescence area was measured, and the density of each image was integrated using Image-Pro Plus 6.0 software to analyze the relative staining level of each image while ensuring consistency in parameters [36].
Enzyme-linked immunosorbent assay (ELISA)
The serum or supernatant collected from each group of mice or NP cells was subjected to ELISA using the ELISA assay kit (Elabscience, Wuhan, China) according to the manufacturer’s instructions to detect the expression of IL-6, iNOS, TNF-α, and IL-1β. The kit numbers used were E-EL-M0044, E-EL-M0696, E-EL-M3063, and E-EL-M0037. In brief, standard samples and specimens were transferred to individual wells and incubated to allow protein binding with immobilized antibodies. After washing, biotinylated specific antibodies were added. After a certain incubation period, unbound biotinylated antibodies were washed away, followed by the addition of HRP conjugate and subsequent washing. 3,3’,5,5’-Tetramethylbenzidine (TMB) substrate was added, turning the solution blue, which further turned yellow upon the addition of the stop solution. The optical density (OD) values were measured at 450 nm after subtracting the blank well values. The expression of each inflammatory factor was calculated based on the protein concentration curve of standard samples [45].
Cell viability assay using CCK-8
The NP cells to be tested were digested and resuspended, and the cell concentration was adjusted to 1 × 105 cells/mL. The cells were seeded in a 96-well plate with 100 µL per well and incubated overnight. Cell viability was determined using the CCK-8 assay kit (C0041, Beyotime, Shanghai, China) according to the manufacturer’s instructions at 12, 24, 36, and 48 h after incubation. For each measurement, 10 µL of CCK-8 detection reagent was added and incubated at 37℃ with 5% CO2 for 1 h. The absorbance at 450 nm was then measured using an enzyme-linked immunosorbent assay reader to calculate cell viability [46].
Edu staining
The NP cells to be tested were seeded in a 24-well plate with a density of 1 × 105 cells per well, and each cell group was replicated in 3 different wells. A solution of 5-Ethynyl-2’-deoxyuridine (Edu) at 10 µmol/L was added to the culture medium from ST067 (Beyotime, Shanghai, China). The plate was then incubated in a cell culture incubator for 2 h. The culture medium was removed, and the cells were fixed with a PBS solution containing 4% paraformaldehyde for 15 min at room temperature. Subsequently, the cells were washed twice with PBS containing 3% BSA. A PBS solution containing 0.5% Triton-100 was added, and the cells were incubated at room temperature for 20 min. After two washes with PBS containing 3% BSA, 100 µL of the staining solution was added to each well, and the plate was incubated at room temperature in the dark for 30 min. DAPI was then added to stain the cell nuclei for 5 minutes. Following that, 6–10 random fields of view were observed under a fluorescence microscope (FM-600, Shanghai Putian Optical Instrument Co., Ltd.), and the number of positive cells in each field was recorded. The Edu labeling rate (%) was calculated as the number of positive cells divided by the sum of positive and negative cells multiplied by 100% [46]. Each experiment was performed in triplicate.
Transwell migration experiment
After various treatments, the NP cells were subjected to a Transwell migration assay. First, the NP cells were diluted in basal culture medium without FBS to a 2.5 × 104 cells/mL. Then, 100 µL of the cell suspension was added to the upper chamber of each Transwell, while 500 µL of medium containing 10% FBS was added to the lower chamber. After 24 h, the upper chamber was removed, and cells in the upper chamber were gently removed using a cotton swab. Next, the cells were fixed with a solution containing 4% paraformaldehyde at room temperature for 30 min. Subsequently, the cells were stained with 0.1% crystal violet (C0121, Beyotime, Shanghai, China) for 30 min. Five random areas were selected, and the cells were photographed and counted under an inverted microscope (IXplore Pro, Olympus, Japan) [47]. Each experiment was performed in triplicate.
Scratch experiment
On the bottom surface of a 6-well plate, evenly draw lines at intervals of 0.5–1 cm using a ruler and a marker. Each hole should be intersected by at least 5 lines. NP cells were seeded into the wells at a density of 5 × 105 cells per well and allowed to grow until reaching 100% confluence. A scratch was made using a 200 µL pipette tip perpendicular to the marked lines, followed by a medium change to serum-free culture medium. The distance between the wound edges was measured and recorded under an optical microscope (model: DM500, Leica) at 0 h and 24 h. Images of the cell groups were captured using an inverted microscope to observe their migration ability. Image-Pro Plus 6.0 was used to analyze the distance between the scratches, and the wound healing rate was calculated according to the following formula [48].
In this study, the variables distance0h and distance24h represent the distances between scratched cells at 0 h and 24 h after the scratch, respectively.
Knockout of CFIm25 using CRISPR-Cas9 technology
CFIm25-KO cells were generated using the CRISPR/Cas9 technology. The sgRNA targeting the Nudt21 gene was designed using the CRISPOR online tool. The sgRNA sequences used were as follows: Nudt21-sgRNA: Forward: 5’-TCTCGACTGTTAATGGCGGG-3’ (PAM: TGG), Reverse: 5’-AAGAGGCGCGGAACACACGC-3’ (PAM: CGG).
The sgRNA was inserted into the Lenti-CRISPR v2 vector (HanBio, Shanghai, China) containing the Streptococcus pyogenes Cas9 nuclease gene. Cell transduction was performed using the lentiviral Lenti-CRISPR v2 vector, followed by the generation of CFIm25-KO cells using the CRISPR/Cas9 editing system.
Transfected cells, including the sgRNA plasmid and donor sequence, were selected with 4 µg/mL puromycin (HY-K1057, MedChemExpress, USA). Surviving cells were then subjected to limited dilution cloning, and CFIm25-KO cells were selected through RT-qPCR and Western blot analysis [45, 49].
Overexpression of CFIm25 using lentivirus
The plasmid vector pCMV6-AC-GFP (LM-2069, LMAI Bio, Shanghai, China) was used by LMAI Bio (Shanghai, China) to construct the CFIm25 plasmid. To generate CFIm25-UP lentivirus (CFIm25-UP-LTEP-s, hereafter referred to as CFIm25-UP) and control lentivirus (NC-LTEP-s, hereafter referred to as Mock), HEK293T cells (Bio-72947, Beijing Biodex Biotechnology Co., Ltd.) were employed. Plasmids and lentivirus packaging services were provided by LMAI Bioengineering. The constructed luciferase reporter gene plasmids (Mock-luc, CFIm25-UP-luc), along with the helper plasmid, were co-transfected into HEK293T cells using Lipofectamine 2000 reagent (11668030, Thermo Fisher, USA). The transfected cells were assessed, amplified, and purified to obtain packaged lentivirus.
For lentiviral-mediated cell transduction, 5 × 105 cells were seeded in a 6-well plate. Once the NP cell confluency reached 60–70%, the medium containing an appropriate amount of packaged lentivirus (MOI = 10, with a working titer of approximately 5 × 106 TU/mL) and 5 µg/mL polybrene (TR-1003, Sigma-Aldrich, UK) was added for transduction. After 4 h of transduction, an equal medium volume was added to dilute polybrene. Following a 24-hour transduction period, fresh medium was replaced. After 48 h of culturing, the transduction efficiency was assessed using the luciferase reporter gene, and the cells were further maintained in a medium supplemented with 2 µg/mL puromycin (E607054, LMAI Bioengineering, Shanghai, China). During cell passaging, the puromycin concentration was gradually increased in a gradient of 2, 4, 6, 8, and 10 µg/mL to select resistant cell lines. Once the cells no longer died in a medium containing puromycin, they were collected, and the knockout efficiency was validated using RT-qPCR [50].
The cell groups were as follows: CFIm25-WT NP cells (wild-type cells), CFIm25-KO NP cells (CFIm25 knockout cells), CFIm25-UP NP cells (CFIm25 overexpression cells), and Mock NP cells (cells transfected with empty lentivirus).
Detection of gene expression by RT-qPCR
Total RNA from tissues and cells was extracted using the Trizol Reagent kit (A33254, Thermo Fisher, USA). Reverse transcription was performed using the reverse transcription kit (RR047A, Takara, Japan) to obtain the corresponding cDNA. The reaction system was prepared using the SYBR® Premix Ex TaqTM II kit (DRR081, Takara, Japan) and the RT-qPCR reaction was carried out in a real-time fluorescence quantitative PCR instrument (ABI7500, Thermo Fisher, USA). The PCR program was designed as follows: an initial denaturation at 95 °C for 30 s, followed by a cycling stage consisting of denaturation at 95 °C for 5 s, annealing at 60 °C for 30 s, and 40 cycles. Subsequently, the extension was performed at 95 °C for 15 s and 60 °C for 60 s, followed by a final extension at 90 °C for 15 s to generate amplification curves. The housekeeping gene, GAPDH, was used as an internal reference. Each RT-qPCR was set up in triplicates, and the experiment was repeated three times. The fold change in gene expression between the experimental and control groups was calculated using the 2−ΔΔCt method, where ΔΔCT = ΔCt experimental group - ΔCt control group, and ΔCt = Ct target gene - Ct reference gene. Ct represents the number of amplification cycles required for the real-time fluorescence intensity to reach the set threshold, at this point, the amplification enters the exponential phase [45]. Primer design details can be found in Table S1.
Western blot analysis
To extract total protein from tissue and cell lysates, RIPA buffer (P0013B, Beyotime, Shanghai, China) containing 1% PMSF (phenylmethanesulfonyl fluoride) was used following the manufacturer’s instructions. The protein concentration of each sample was determined using the BCA assay kit (P0011, Beyotime, Shanghai, China). The protein concentration was then adjusted to 1 µg/µL, and each sample was set at a volume of 100 µL. Samples were boiled at 100 °C for 10 min to denature the proteins and stored at -80 °C until further use.
An 8-12% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) was prepared based on the target protein band size. An equal amount of 50 µg protein sample was loaded onto each lane using a microsyringe. Electrophoresis separation was performed at a constant voltage of 80 V for 2 h, followed by 120 V for an additional 90 min at a constant current of 250 mA. The proteins from the gel were then transferred to a PVDF membrane (1620177, Bio-Rad, USA).
The membrane was blocked at room temperature for 1 h with 1× TBST (Tris-buffered saline with Tween 20) containing 5% skim milk. After discarding the blocking solution, the membrane was washed with 1× TBST for 10 min. The primary antibody (refer to Table S2 for antibody information) was incubated overnight at 4 °C, followed by three washes with 1× TBST for 10 min each. Subsequently, the membrane was washed three times with 1× TBST at room temperature for 5 min each. HRP-conjugated goat anti-rabbit IgG (ab6721, dilution: 1:5000, Abcam, Cambridge, UK) or goat anti-mouse IgG (ab205719, dilution: 1:5000, Abcam, Cambridge, UK) secondary antibody was added and incubated at room temperature for 1 h. After three washes with 1× TBST at room temperature for 5 min each, the membrane was immersed in an ECL reaction solution (1705062, Bio-Rad, USA) and incubated at room temperature for 1 min. The liquid was removed, and the membrane was covered with plastic wrap for band exposure using the Image Quant LAS 4000 C gel imaging system (GE, USA). The relative protein expression levels were quantified by comparing the grayscale value ratio of the target band to the reference band, with GAPDH serving as an internal control for total cellular protein. Protein expression levels were analyzed for each protein [45]. Each experiment was repeated three times.
Preparation of GelMA-HAMA composite hydrogel
Synthesis of Methacrylate Gelatin (GelMA): Initially, a 10% (w/v) phosphate-buffered solution with a pH of 7.4 was prepared using type A gelatin (G2500, Sigma-Aldrich, UK) derived from pig skin. Subsequently, a solution of 20% methacrylic anhydride (MA, 276685, Sigma-Aldrich, UK) was added dropwise to the prepared gelatin solution at 50 °C and stirred for 1 h. The resultant mixture was then diluted 5-fold with a 40 °C heated phosphate-buffered solution, filtered, and dialyzed using distilled water for 1 week before freeze-drying (Figure S7A) [32].
Synthesis of Methacrylate Hyaluronic Acid (HAMA): Initially, a distilled water solution of hyaluronic acid (924474, Sigma-Aldrich, UK) was prepared at a concentration of 0.5% (w/v). Dimethylformamide (DMF) was added to the solution at a ratio of H2O: DMF = 3:2. While stirring, a 1% solution of MA (276685, Sigma-Aldrich, UK) was added dropwise. The pH was then adjusted to 8–9 using 0.5 M sodium hydroxide (S5881, Sigma-Aldrich, UK), and the solution was incubated overnight with continuous stirring at 4 °C. Following this, the solution was dialyzed using distilled water for 3 days and subsequently freeze-dried (Figure S7B) [32].
Preparation of GelMA-HAMA Microspheres: HAMA and GelMA were mixed in a 3:1 ratio, followed by the addition of 0.3% (w/v) photoinitiator (L157759, Aladdin, Shanghai, China), mineral oil (ST275, Beyotime, Shanghai, China), and 10% (w/w) Span80 (1338-43-8, Macklin, Shanghai, China) as a surfactant. Crosslinking of the hydrogel was achieved through UV irradiation (365 nm, 6.9 mW·cm− 2) in a dark setting. The flow rate was adjusted using a microfluidic device to prepare GelMA-HAMA microspheres. The collected microspheres were washed repeatedly with isopropanol and 75% ethanol to remove surfactants and mineral oil. The purified microspheres were then soaked in PBS for 4 h, repeated 6 times, to eliminate other additives. To obtain porous microspheres, the purified spheres were frozen at -20 °C for 8 h and subsequently freeze-dried for 48 h [35, 51].
Preparation of GelMA-HAMA microspheres
GelMA-HAMA microspheres conjugated with APET×2 polypeptide (GelMA-HAMA-APET×2, GA): Disperse 100 mg of GelMA-HAMA microspheres in 1 mL of MES buffer (pH = 6, HY-D0858, MedChemExpress, Shanghai, China). Then sequentially add 8 mg of EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, 22980, Thermo Fisher, USA) and 12 mg of NHS (N-hydroxysuccinimide, 24500, Thermo Fisher, USA) and react at 37 ℃ for 15 min. Subsequently, add 5 µg of APET×2 (ab141849, Abcam, UK) and incubate at 37 ℃ for 6 h. Rapid centrifugation yields GA, which is washed three times and then freeze-dried for 48 h to obtain GA hydrogel [35].
GCA hydrogel loaded with Cas9-CFIm25-sgRNA recombinant protein (GelMA-HAMA-APET×2@ Cas9-CFIm25-sgRNA, GCA): Dissolve Cas9-CFIm25-sgRNA recombinant protein at a concentration of 2 mg/mL in PBS and mix it with 10% (wt) GA hydrogel. Incubate at 37 ℃ for 30 min, then wash three times, centrifuge, and dry to obtain GCA [25, 52].
Characterization of composite hydrogels
The composite hydrogel was characterized and detected using Fourier Transform Infrared spectroscopy (FTIR) and Scanning Electron Microscopy (SEM). Briefly, SEM analysis was conducted using a Hitachi S-4800 SEM with an acceleration voltage of 3 kV. The hydrogel sample was placed on conductive tape and coated with gold using Cressington Scientific Instruments’ coating equipment in Watford, UK, for 60 s. Subsequently, the sample was observed using the SEM.
For the FTIR analysis, the hydrogel was frozen in a -20℃ freezer and freeze-dried for 48 h. The freeze-dried sample (1 mg) was mixed with 80 mg of dried KBr (221864) from Sigma, ground into a fine powder, and pressed into a pellet. The pellet was then scanned using the Nicolet 6700 infrared spectrometer from Thermo Fisher, with specific parameters for single-beam absorption mode (resolution: 4 cm− 1, wavelength range: 4000 –400 cm− 1, scan speed: 0.15 cm/s, number of scans: 128). During the scanning process, the sample was purged with pure helium at a flow rate of 5 mL/min to remove air.
In the in vitro release experiment, the GCA hydrogel solution was dissolved in 1 mL of PBS and incubated at 37℃ with 5% CO2. The supernatant was collected every 2 days and replaced with fresh PBS. The released protein quantity in the culture medium was determined using the Bradford protein assay kit from Shanghai Biotehnai Co., Ltd., following the instructions provided in the kit. [32, 35].
In vivo animal experiment
A total of 24 male C57BL/6J mice were involved in the experiment and were randomly divided into two groups. One group consisted of 6 mice, while the other group consisted of 18 mice. The IDD model was constructed in 18 mice using the method described above, and they were randomly divided into 3 groups, with 6 mice in each group. The remaining 6 mice were exposed to the intervertebral disc area but were not induced by IDD. These mice were divided into 4 groups and received different treatments: the Sham group, where intervertebral disc exposure was performed without injury, and 10 µL of PBS was injected into the intervertebral disc as treatment; the model control group (PBS group), where mice were injected with 10 µL of PBS after inducing IDD; the treatment group (GA group), where mice were injected with 10 µL of GA solution into the intervertebral disc; and the treatment group (GCA group), where mice were injected with 10 µL of GCA solution into the intervertebral disc. Drug treatment was initiated one week after constructing the intervertebral disc model, with injections administered every 2 days for a duration of 4 weeks. After completing the treatment, the mice were subjected to radiographic examination and euthanized, and their intervertebral disc tissues were extracted for subsequent biochemical experiments [35].
Proteomic analysis
Six mice were randomly selected and divided into two groups, the PBS group (n = 3) and the GCA group (n = 3), for injection therapy. The protein from the intervertebral disc NP tissue of mice was extracted using RIPA buffer supplemented with a protease inhibitor (Figure S3). During the extraction process, samples were treated with an ultrasound probe for 30 s every 5 min, three times in total, to ensure cell rupture and protein release. The protein concentration of the extracted samples was determined using the BCA protein quantification kit, and the concentration was ensured to be within an acceptable range. After adjusting the pH to 8.0, trypsin (15090046, Thermo Fisher, USA) was added at a ratio of 1:50 (enzyme to protein), and the mixture was incubated at 37 °C for 16 h for enzymatic digestion. The digested samples were then subjected to clean-up using ZipTip C18, loaded onto a high-performance liquid chromatography system, and connected to a mass spectrometer for MS/MS analysis. The data were processed using the MaxQuant software, which included protein identification and quantification.
iTRAQ labeling was performed using desalted peptides containing 0.1% formic acid (1002641000, Sigma, USA) for mass spectrometry analysis. Each sample was analyzed three times using the QSTAR Elite Hybrid MS (Applied Biosystems/MDS-SCIEX) and an online HPLC system (Shimadzu, Japan). In each analysis, 30 µL of peptide solution was injected and separated on a homemade nano-column with a nano-electrospray emitter (75 μm ID × 15 cm, 5 μm particle size) (New Objectives, Woburn, MA). A 90-minute high-performance liquid chromatography gradient was established using mobile phase A (5% (v/v) acetonitrile, 0.2% (v/v) formic acid) and mobile phase B (90% (v/v) acetonitrile, 0.2% (v/v) formic acid), with an effective flow rate of 300 nL/min. The separations were performed using a constant flow rate of 30 µL/min. The mass spectrometer was set to acquire data in positive ion mode, with a mass range of 300–2000 m/z and precursor fragmentation for charges + 2 to + 4. The three most abundant peptide ions with a count threshold greater than 5 were selected in each MS/MS spectrum. The dynamic exclusion time for selected precursor ions was set to 30 s, with a mass tolerance of 30 mDa.
LC-MS/MS injections were performed utilizing automated collision energy and automatic mass spectrometry/mass spectrometry accumulation smart information dependency. The fragmentation intensity multiplier was set to 20, with a maximum accumulation time of 2 s. Three LC-MS/MS injections (technical replicates = 3) were carried out to achieve better coverage of the target proteome and improve statistical consistency. WB analysis was used for protein verification after proteomics analysis of the same sample. The parameters were as follows: (1) MS: scan range (m/z) = 350–1500; resolution = 120,000; AGC target = 4e5; maximum injection time = 50 ms; (2) HCD-MS/MS: resolution = 30,000; AGC target = 1e5; collision energy = 33; (3) DIA (data-independent acquisition). Each window overlapped by 1 m/z, and the window number was 47. The iRT kit (Ki3002, Biognosys AG, Switzerland) was added for calibration of peptide peak retention times. The DIA dataset was generated by Spectronaut V 13 (Biognosys AG, Switzerland), including data normalization and relative protein quantification. Differential expression proteins were filtered using Welch’s ANOVA test with a filtering condition of p-value < 0.05 and logFC > 1.0 [53].
TUNEL detection of cellular apoptosis
Tissue cells were stained using the TUNEL staining kit (C1088, Beyotime, Shanghai, China). Briefly, mouse intervertebral disc tissue was fixed with 4% paraformaldehyde for 30 min, followed by three washes with PBS. Then, a permeabilization solution containing 0.3% Triton X-100 in PBS was added and incubated for 3 min at room temperature, followed by 5 min at room temperature. This was followed by two additional washes with PBS. Subsequently, 50 µL of TUNEL detection solution was added, and the samples were incubated in the dark at 37 °C for 60 min. After incubation, three washes with PBS were performed, followed by a 10-minute restaining with DAPI (10 µg/mL), and finally, the slides were sealed using an anti-fluorescence quenching mounting solution. Cellular apoptosis was observed using a fluorescence microscope, with Cy3 exhibiting an excitation wavelength of 550 nm and an emission wavelength of 570 nm (red fluorescence). The apoptotic cell proportion in each group was calculated using Image-Pro Plus 6.0 software [36].
Flow cytometry analysis
Initially, cells were isolated from mouse intervertebral disc tissue samples and treated with collagenase and DNAse for approximately 1–2 h to obtain a single-cell suspension using density gradient centrifugation. Subsequently, the cells were adjusted to approximately 1 × 106 cells/mL and incubated for 5 h in a stimulant solution containing PMA (Phorbol 12-myristate 13-acetate, HY-18739, MedChemExpress, Shanghai, China), ionomycin (HY-13434, MedChemExpress, Shanghai, China), and GolgiPlug™ (Brefeldin A, 555029, BDbiosciences, USA) at 37 °C and 5% CO2. After incubation with CD16/CD32 antibodies (0.5 mg/mL, 14-0161-82, Thermo Fisher, USA) for 10 min to block nonspecific binding, surface labeling was performed as follows: APC-conjugated CD19 antibody (0.2 mg/mL, 17-0193-82, Thermo Fisher, USA) for B cells, PE-conjugated CD3 antibody (0.2 mg/mL, 12-0031-82, Thermo Fisher, USA) for T cells, FITC-conjugated CD4 antibody (0.5 mg/mL, 11-0041-82, Thermo Fisher, USA) for CD4+ T cells, PE-Cy7-conjugated CD8 antibody (0.2 mg/mL, A15385, Thermo Fisher, USA) for CD8+ T cells, APC-conjugated antibody for IFN-γ (0.2 mg/mL, 17-7311-82, Thermo Fisher, USA) for Th1 cells, and PE-conjugated antibody for IL-4 (0.2 mg/mL, 12-7041-82, Thermo Fisher, USA) for Th2 cells. Cell analysis was carried out using a flow cytometer, ensuring differentiation of cell types by setting appropriate gates and employing unstained cells and singly stained controls to verify staining specificity and accuracy. All procedures were conducted under sterile conditions to prevent cell contamination [54].
Statistical analysis
Data were obtained from at least three independent experiments and presented as the mean ± standard deviation (Mean ± SD). For comparisons between the two groups, an independent-sample t-test was used. For comparisons among three or more groups, a one-way analysis of variance (ANOVA) was employed. If the ANOVA results indicated significant differences, Tukey’s HSD post hoc test was further performed to compare the differences between each group. For non-normally distributed or heterogeneous variance data, the Mann-Whitney U test or Kruskal-Wallis H test was used. All statistical analyses were conducted using GraphPad Prism 9.5.0 (GraphPad Software, Inc.) and R version 4.2.1 (R Foundation for Statistical Computing). The significance level for all tests was set at 0.05, with a two-sided p-value less than 0.05 considered statistically significant, while a p-value greater than 0.05 was deemed non-significant.
Results
Single-cell RNA sequencing reveals dynamic changes in cell types during IDD
We first established the IDD mouse model by using a puncture needle (Figure S4A) and evaluated the intervertebral disc through imaging and histological sectioning after 4 weeks. X-ray examination revealed a reduced intervertebral disc index and decreased intervertebral disc height in the IDD group compared to the Sham group, serving as the control group (Figure S4B). HE staining showed structural damage to the intervertebral disc in the IDD model mice, with rupture of the NP tissue and disruption of the boundary between the NP and the fibrous ring (Figure S4C). Safranin O-Fast Green staining further confirmed tissue damage in the intervertebral disc of the IDD model mice, with decreased red staining indicating a decrease in cartilage components and increased green staining suggesting an increase in bone components (Figure S4D). The histological score of the intervertebral disc in IDD mice was significantly higher, indicating significant degeneration of the intervertebral disc (Figure S4E). Additionally, immunohistochemical staining revealed a significant decrease in the content of collagen type II (COLII) and aggrecan in the intervertebral disc tissue of the IDD group compared to the Sham group (Figure S4F). These findings demonstrate our successful construction of the IDD mouse model.
To further analyze changes in intervertebral disc cell populations during IDD development and identify the molecular mechanisms involved, we collected samples of degenerative intervertebral disc tissue at 2, 4, and 8 weeks post-surgery (referred to as the 2 wks group, 4 wks group, and 8 wks group, respectively), as well as healthy intervertebral disc tissue from normal mice (Sham group). scRNA-seq was performed to analyze these samples, and the data were integrated using the “Seurat” package in the R programming language (Figure S1). First, we examined the gene count (nFeature_RNA), mRNA molecule count (nCount_RNA), and percentage of mitochondrial genes (percent.mt) in all cells of the scRNA-seq data. The majority of cells had nFeature_RNA < 5000, nCount_RNA < 20,000, and percent.mt < 20% (Figure S5A). Based on these criteria, we excluded low-quality cells, resulting in an expression matrix of 15,993 genes and 13,301 cells. The correlation analysis of sequencing depth showed a correlation coefficient of r = -0.2 between nCount_RNA and percent.mt and r = 0.9 between nCount_RNA and nFeature_RNA in the filtered data (Figure S5B). This indicates that the filtered cell data has good quality and can be used for further analysis.
Next, we conducted further analysis on the filtered cells. Firstly, highly variable genes were selected based on gene expression variance, and the top 2000 genes with the highest variance were chosen for downstream analysis (Figure S5C). Cell cycle scores were then calculated using the CellCycleScoring function (Figure S5D), and the data was preliminarily normalized. Subsequently, PCA was performed to linearly reduce the dimensionality of the data, as depicted in Figure S5E, which shows the first two principal components. The expression profiles of genes most correlated with these principal components were visualized in a heatmap (Figure S5F), and the distribution of cells in the PC1 and PC2 space was represented in Figure S5G. The results demonstrated the presence of batch effects between samples.
To alleviate batch effects and improve the accuracy of cell clustering, we employed the harmony package for batch correction of the sample data (Figure S5H). Additionally, an Elbow Plot was used to visually assess the standard deviation ordering of principal components (PCs). The plot revealed that PC1 to PC20 adequately reflected the information of the selected highly variable genes and held significant analytical significance (Figure S5I). The batch effects were effectively eliminated after correction, as observed in Fig. 1A.

Cell clustering of scRNA-seq data. Note (A) Distribution of cells after batch correction in PC_1 and PC_2. Each point represents a cell; (B) UMAP visualization of cell clustering, displaying the clustering and distribution of cells from the Sham group, IDD postoperative 2 weeks, 4 weeks, and 8 weeks samples. Late Sham samples are depicted in purple, IDD postoperative 2 weeks samples in red, IDD postoperative 4 weeks samples in green, and IDD postoperative 8 weeks samples in cyan; (C) UMAP visualization of cell clustering, providing an overall representation of the clustering and distribution of cells from different sources, with each color representing a cluster; (D) Visualization of cell clustering and distribution based on different source samples, with each color representing a cluster; (E) UMAP visualization of cell annotation results based on cell clustering, with each color representing a cell subtype; (F) Expression levels of 6 cell marker genes in different cell subtypes, with darker green indicating higher average expression levels
Furthermore, the UMAP algorithm was utilized for non-linear dimensionality reduction of the top 20 principal components. Through UMAP clustering analysis, all cells were grouped into 10 distinct cell clusters (Fig. 1B-D). Automatic annotation of these 10 cell clusters was performed using the Bioconductor/R package “SingleR,” resulting in the identification of 6 cell types: chondrocytes, immune cells (IC), macrophages (Mac), stromal cells (SC), pericytes, and endothelial cells (EC) (Fig. 1E). The UMAP expression patterns of marker genes representing these 6 cell types were displayed, with Dmp1 representing chondrocytes, Fcer1g associated with IC, Abhd12 assigned to Mac, CD18 representing SC, CD146 reflecting pericytes, and CD105 indicating EC (Fig. 1F). Additionally, a heatmap was generated showcasing the top 5 genes for each cell type (Figure S5J).
In summary, our scRNA-seq analysis successfully identified 6 cell types within the IVD tissue.
Key roles of chondrocytes and immune cells in IDD and upregulation of CFIm25 expression
In this study, we provide a detailed description of the cellular composition of six types of cells from four samples. By performing a T-test, we analyzed the differences in cell quantities between the Sham group and the IDD group at 2, 4, and 8 weeks post-surgery. The analysis revealed a significant decrease in chondrocytes and a significant increase in immune cells and macrophage quantities in the IDD group at 2, 4, and 8 weeks (Fig. 2A).

Identification of cell-cell interactions in IDD NP tissue cells. Note (A) Bar plot showing the overall cell composition proportions in the Sham group and IDD postoperative 2, 4, and 8 weeks samples, with cell types color-coded as IC (immune cells), Mac (macrophages), SC (stromal cells), and EC (endothelial cells); (B) Circos plot displaying the number of cell communications in the Sham group and IDD postoperative 2, 4, and 8 weeks groups, with the thickness of the lines indicating the number of pathways; (C) Circos plot showing the number of cell communications between chondrocytes and other cells in the Sham group and IDD postoperative 2, 4, and 8 weeks groups, with the thickness of the lines indicating the number of pathways; (D) Display of ligand-receptor pairs significantly associated with chondrocytes in the Sham group and IDD postoperative 2, 4, and 8 weeks groups
To further investigate the mechanisms driving cellular evolution in IDD, we examined the intercellular signaling mediated by ligand-receptor interactions. We used the “CellChat” package in R to infer the communication and interaction levels between identified phenotypes in degenerated intervertebral disc cells. The results demonstrated a significant increase in both the number of cell-cell communications and interaction strengths among cell subpopulations at 2 weeks post-IDD (Fig. 2B). Additionally, we observed a significant increase in cell-cell communication and interaction strengths, particularly between chondrocytes and immune cells, following IDD (Fig. 2C). These findings underscore the importance of chondrocytes in the IDD model. Furthermore, we identified an increase in the number of cell-cell communications and receptor-ligand pairs between chondrocytes and immune cells post-IDD, indicating that chondrocytes interact with immune cells to enhance the inflammatory response and influence the function of the intervertebral disc (Fig. 2C-D). Altogether, these results highlight the critical role of chondrocytes and immune cells in mediating intercellular communication during IDD.
To further explore the mechanisms regulating IDD, we performed transcriptome sequencing (RNA-seq) on IDD samples. A total of 1,408 differentially expressed genes were identified, including 840 upregulated genes and 568 downregulated genes (Fig. 3A). Additionally, we selected 308 differentially expressed genes, specifically in chondrocytes from the NP and found an overlap of 122 genes between these two sets (Fig. 3B). For these 122 overlapping genes, we conducted PPI analysis using STRING, obtaining the PPI network and interaction rankings of the genes. Figure 3C-D provides detailed information, highlighting the central role of the Nudt21 gene in this protein interaction network.

Identification of key genes mediating IDD through scRNA-seq and transcriptome sequencing analysis. Note (A) Volcano plot showing the differentially expressed genes between normal IVD NP tissue from three groups of mice and degenerative IVD NP tissue from three groups of mice. Yellow upward triangles represent upregulated genes, green downward triangles represent downregulated genes, and black dots represent non-differentially expressed genes; (B) Venn diagram showing the intersection between RNA-seq differentially expressed genes and scRNA-seq differentially expressed genes; (C) Statistics of interaction sites in the PPI network of proteins encoded by the 122 intersecting genes, shown graphically; (D) PPI interaction network diagram of the proteins encoded by the 122 intersecting genes (combined score = 0.7), with color gradient indicating the degree value of the genes from small to large; (E) Bubble plot showing the GO enrichment analysis of the 122 intersecting genes, with circle color representing the significance of enrichment and circle size representing the number of enriched genes; (F) Bubble plot showing the KEGG enrichment analysis of the 122 intersecting genes, with circle color representing the significance of enrichment and circle size representing the number of enriched genes; (G) LASSO algorithm selected 5 feature genes; (H) SVM-RFE algorithm selected 3 feature genes; (I) Venn diagram showing the intersection of the two machine learning results, resulting in 1 gene; (J) scRNA-seq detection of Nudt21 gene expression; (K) RT-qPCR detection of Nudt21 mRNA expression in mouse IVD NP tissue (n = 3); (L) Western blot detection of CFIm25 protein expression in mouse IVD NP tissue (n = 6). Quantitative data in the figures are represented as Mean ± SD, with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, indicating statistical significance compared to the Sham group
In addition, we conducted a GO functional enrichment analysis on the 122 intersecting genes. The results revealed that these intersecting genes were associated with various biological processes, such as muscle contraction, salt response, and muscle cell development, among others. In terms of cellular components, they were mainly enriched in items related to muscle fibers, contraction fibers, and muscle segments. Regarding molecular function, the main enrichment was observed in items related to muscle actin binding, endopeptidase inhibitor activity, and peptide inhibitor activity (Fig. 3E). Furthermore, we performed KEGG pathway enrichment analysis on the 122 intersecting genes, and the results showed that these 76 EARGs were primarily involved in the IL-17 signaling pathway, mRNA surveillance pathway, TNF signaling pathway, pertussis, and typhoid pathway, among others (Fig. 3F).
Next, we employed machine learning techniques to screen the 122 intersecting genes. We utilized LASSO regression modeling and the SVM-RFE algorithm to identify the feature genes associated with IDD. In the LASSO regression algorithm, we selected the Lambda parameter and determined the point of minimum error through cross-validation, resulting in the identification of five key genes (Fig. 3G). In the SVM-RFE algorithm, we used the “svmRadial” method to select three key genes (Fig. 3H). The intersection of genes obtained from both algorithms was visualized in a Venn diagram, and the feature gene Ndut21 was identified (Fig. 3I). Additionally, in our single-cell UMAP plot, we observed higher expression of the Ndut21 gene in the intervertebral disc NP tissue (Fig. 3J). Therefore, we speculate that the Ndut21 gene may play a significant role in regulating IDD.
To determine the role of Ndut21 in IDD further, we established an IDD mouse model through surgery and validated the expression of Ndut21 in normal and degenerated intervertebral disc NP tissues using RT-qPCR and Western blot. The results showed a significant increase in Ndut21 mRNA and its encoded protein CFIm25 expression in the IDD group compared to the Sham group, and the expression levels of Ndut21 mRNA and CFIm25 protein gradually increased with the progression of IDD (Fig. 3K-L).
Taken together, our findings from the multi-omics analysis of IDD in the NP tissue highlight the importance of chondrocytes and immune cells as key mediators of intercellular communication in IDD. The mRNA and protein expression levels of CFIm25 were significantly upregulated in the degenerated tissues, suggesting their potential involvement in the physiological and pathological processes of intervertebral disc cells.
The role of CFIm25 in regulating function and immune response in intervertebral disc NP cells
To investigate the impact of CFIm25 on IDD further, we isolated NP cells from mouse intervertebral disc NP tissue and subjected them to an in vitro culture. Verification was performed through immunofluorescent staining of COLII protein, Safranin O-Fast Green Staining, and alcian blue staining. The results of these tests demonstrated the successful isolation of NP cells from mouse IVD, with an expression of COLII protein, alkaline cartilage components, and proteoglycan (Figure S6A).
To mimic the degenerative environment, we simulated degenerated NP cell models by adding pro-inflammatory cytokines such as TNF-α and IL-1β, which resulted in degenerated NP cells (Figure S6B-C).
Alcian blue staining results revealed a significant decrease in proteoglycan expression in degenerated NP cells (Degeneration group) compared to normal NP cells (Normal group) (Figure S6D). Expression of COLII and Aggrecan proteins in degenerated NP cells was also significantly reduced (Figure S6E), while expression of inflammatory factors IL-6 and iNOS was significantly increased (Figure S6F). Additionally, CCK-8 and EDU assays demonstrated decreased viability (Figure S6G) and significantly reduced proliferative capacity (Figure S6H) in degenerated NP cells compared to normal NP cells. Transwell migration and cell scratch experiments further revealed a significant decrease in the migration ability of degenerated NP cells (Figure S6I-J). Moreover, the expression of CFIm25 was significantly increased in degenerated NP cells compared to normal NP cells (Figure S6K).
Based on degenerated NP cells, we performed CRISPR-Cas9 knockout of CFIm25 or overexpression of CFIm25 through lentiviral plasmid transfection, resulting in CFIm25 knockout and overexpression of degenerated NP cell lines (Fig. 4A). The effects of gene knockout and overexpression were confirmed by Western blot analysis (Fig. 4B). EDU detection results demonstrated a significant enhancement in the proliferation ability of degenerated NP cells in the CFIm25-KO group compared to the CFIm25-WT group, while a significant decrease in proliferation ability was observed in the CFIm25-UP group compared to the Mock group (Fig. 4C). CCK-8 assay results indicated that CFIm25 knockout enhanced cell vitality in degenerated NP cells, whereas CFIm25 overexpression suppressed cell vitality (Fig. 4D). Transwell migration and cell scratch experiments revealed that CFIm25 knockout enhanced the migration ability of degenerated NP cells, while CFIm25 overexpression inhibited cell migration ability (Fig. 4E-F). Additionally, alcian blue staining results showed that CFIm25 knockout increased proteoglycan expression in degenerated NP cells, while CFIm25 overexpression inhibited proteoglycan expression (Fig. 4G). Immunofluorescence staining results demonstrated elevated expression of COLII and Aggrecan proteins in degenerated NP cells of the CFIm25-KO group compared to the CFIm25-WT group, whereas significant downregulation of COLII and Aggrecan protein expression was observed in the CFIm25-UP group compared to the Mock group (Fig. 4H). ELISA revealed an increase in inflammatory factors IL-6 and iNOS levels in degenerated NP cells corresponding to CFIm25 expression levels, while CFIm25 knockout induced a decrease in IL-6 and iNOS expression in degenerated NP cells (Fig. 4I).

Knockdown of the Nudt21 gene suppresses NP cell degeneration Note (A) Flowchart illustrating the knockdown or overexpression of CFIm25; (B) Western blot analysis of CFIm25 protein expression in degenerated NP cells under different interventions; (C) EDU assay to evaluate the proliferative capacity of degenerated NP cells in different intervention groups (scale bar = 25 μm); (D) CCK-8 assay to measure the viability of degenerated NP cells under different interventions at 12, 24, 36, and 48 h; (E) Cell scratch experiment examining the migration ability of degenerated NP cells in different intervention groups (scale bar = 100 μm); (F) Transwell migration assay investigating the migration ability of degenerated NP cells in different intervention groups (scale bar = 50 μm); (G) Alcian blue staining to detect the expression change of proteoglycan in degenerated NP cells under different interventions (scale bar = 50 μm); (H) Immunofluorescence staining to analyze the expression of COLII and Aggrecan proteins in degenerated NP cells in different intervention groups (scale bar = 25 μm); (I) ELISA assay to measure the expression of inflammatory factors IL-6 and iNOS in degenerated NP cells under different interventions. The quantified data in the figures are presented as Mean ± SD, and each experiment was repeated three times. * indicates comparison with the CFIm25-WT group, the significance levels were as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Similarly, compared to the Mock group, the significance levels were: #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001
By constructing an in vitro cell model and conducting knockout and overexpression experiments of CFIm25, we further validated its role in regulating the immune microenvironment. These results not only unveil the multiple functions of CFIm25 in IDD but also provide experimental evidence for further investigating its specific mechanisms in this pathological process.
GelMA-HAMA-APET×2 composite hydrogel promotes proliferation, migration, and differentiation of IDD cells
GelMA and HAMA hydrogel particles were synthesized separately, and GelMA-HAMA microspheres were then synthesized using a photo-initiator (Figure S7A).
APET×2 polypeptide is a selective and reversible acid-sensitive ASIC3 channel inhibitor that reduces the inflammatory factors storm caused by an acidic environment in the intervertebral disc [35]. Carboxyl groups on the surface of GelMA-HAMA microspheres were activated using EDC/NHS and then covalently connected to the amino residues on APET×2, forming GelMA-HAMA-APET×2 microspheres (GA) (Figure S7A). Finally, GelMA-HAMA-APET×2 microspheres were co-cultured with Cas-9-CFIm25-sgRNA protein, constructing GelMA-HAMA-APET×2@Cas-9-CFIm25-sgRNA composite hydrogel microspheres (GCA) (Figure S7A).
The GelMA-HAMA hydrogel with double-targeted APET×2 polypeptide and sgRNA, loaded in a new porous spherical form, was successfully characterized using SEM and FTIR. The SEM results demonstrated the successful coupling of APET×2 polypeptide with the hydrogel and the successful loading of Cas-9-CFIm25-sgRNA protein by GA (Figure S7B). The FTIR results showed that there was no difference in the wavelength between GA and the hydrogel, but the absorbance peak of the hydrogel was stronger than that of GA, suggesting that the hydrogel had more active functional groups (Figure S7C). Drug release testing of the hydrogel showed that it could stably and slowly release both the polypeptide and Cas-sgRNA protein over an extended period of time (Figure S7D). Furthermore, microscopic observation revealed that the hydrogel microspheres were uniformly dispersed in PBS, with a particle size distribution ranging from 104 to 110 μm, and maintained their shape integrity in PBS (Figure S7E). These results demonstrate the successful construction of the GelMA-HAMA hydrogel loaded with APET×2 polypeptide and Cas-sgRNA.
In vitro cellular experiments were conducted to validate the biological activity of the loaded complex. It was predicted that GCA could release APET×2 polypeptide and Cas-9-CFIm25-sgRNA protein in NP cells, leading to the knockdown of CFIm25 and improvement of IDD (Fig. 5A). Western blot analysis showed that compared to the hydrogel GA without Cas-9-CFIm25-sgRNA protein, GCA effectively suppressed CFIm25 expression in NP cells (Fig. 5B). CCK-8 assay demonstrated that GCA significantly enhanced the viability of degenerated NP cells (Fig. 5C). ELISA results revealed that GCA inhibited the expression of IL-6 and iNOS in degenerated NP cells, thereby suppressing inflammation (Fig. 5D). The enhancement of proliferative capacity in degenerated NP cells by GCA was also significantly stronger than that of GA (Fig. 5E). Additionally, the results of the transwell migration experiment and cell scratch experiment indicated that GCA improved the migratory ability of degenerated NP cells (Fig. 5F-G). Immunofluorescence staining showed that compared to the PBS and GA groups, the expression of COLII and Aggrecan proteins in degenerated NP cells was elevated in the GCA group, approaching the level of normal cells (Normal group) (Fig. 5H). Moreover, alcian blue staining results demonstrated that GCA restored the expression level of proteoglycan in degenerated NP cells (Fig. 5I).

GCA significantly inhibits NP cell degeneration. Note (A) Schematic representation of GCA’s action inside the cell; (B) Western blot analysis of CFIm25 protein expression; (C) CCK-8 assay to measure the viability of NP cells under different interventions at 12, 24, 36, and 48 h; (D) ELISA assay to measure the expression of inflammatory factors IL-6 and iNOS in NP cells; (E) EDU assay to evaluate the proliferative capacity of NP cells (scale bar = 25 μm); (F) Transwell migration experiment investigating the migration ability of NP cells (scale bar = 50 μm); (G) Cell scratch experiment examining the migration ability of NP cells (scale bar = 100 μm); (H) Immunofluorescence staining to analyze the expression of COLII and Aggrecan proteins in NP cells (scale bar = 25 μm); (I) Alcian blue staining to detect the expression change of proteoglycan in NP cells (scale bar = 50 μm). The quantitative data in the figure are expressed as Mean ± SD. * indicates comparison between the two groups, *p < 0.05, *p < 0.01, **p < 0.001, ***p < 0.0001, and each experiment was repeated 3 times per group
Based on the comprehensive results of these experiments, we conclude that the novel porous GelMA-HAMA hydrogel loaded with APET×2 polypeptide and sgRNA exhibits promoting effects on the proliferation, migration, and differentiation of degenerated NP cells, demonstrating its potential application in intervertebral disc repair.
GCA treatment promotes regeneration and improvement of IDD in mice
To explore the promotive effect of GCA on IVD regeneration, we evaluated the in vivo effects of GCA using a mouse IDD model established by needle puncture (Fig. 6A). Imaging and histological analysis are reliable indicators for reflecting IDD or regeneration [35]. We constructed the IDD model and assessed the changes in intervertebral disc height in mice four weeks after treatment using X-ray examination, as shown in Fig. 6B. Compared to the Sham group, mice in the PBS group showed a significant reduction in intervertebral disc height. However, mice in the treatment group exhibited a significant recovery in intervertebral disc height, and the improvement in mice treated with GCA exceeded that of mice treated with GA.

GCA effectively improves IDD in mice. Note (A) Schematic diagram of the process for treating IDD in mice with GCA (n = 6); (B) Representative X-ray images of IVD in mice from different treatment groups (scale bar = 5 mm); (C) H&E staining of IVD in mice from different treatment groups (scale bar = 200 μm); (D) Safranin O-Fast Green staining of IVD in mice from different treatment groups, with green representing bone tissue and red representing cartilage tissue (scale bar = 200 μm); (E) Histological scoring of intervertebral disc tissue in mice; (F) Expression of inflammatory factors IL-6, iNOS, TNF-α, and IL-1β in mouse serum detected by ELISA; (G) TUNEL staining to detect cell apoptosis in the NP tissue of mice (scale bar = 200 μm); (H) Immunohistochemical staining showing the expression of CFIm25 protein in intervertebral disc NP tissue of mice (scale bar = 200 μm); (I) Immunohistochemical staining showing the expression of COLII protein in intervertebral disc NP tissue of mice (scale bar = 200 μm); (J) Immunohistochemical staining showing the expression of Aggrecan protein in intervertebral disc NP tissue of mice (scale bar = 200 μm). The quantitative data in the figure are expressed as Mean ± SD, with 6 mice in each group. * indicates comparison between the two groups, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Histological analysis of the mouse intervertebral disc tissue sections stained with H&E as well as Safranin O-Fast Green staining revealed the following: Compared to the Sham group, mice in the PBS group exhibited ruptured NP, fuzzy boundaries between the NP and annulus fibrosus, and a significant decrease in cartilage components within the tissue. In comparison to the PBS group, mice in the treatment group showed improved intervertebral disc tissue degeneration and structural damage, clearer boundaries between the NP and annulus fibrosus, and reduced loss of cartilage components within the intervertebral disc tissue. Particularly, the improvement in mice with degenerated IVD treated with GCA was more pronounced than that in the GA treatment group (Fig. 6C-D). Additionally, the histological scores of intervertebral disc tissue in the GCA group were significantly decreased, indicating a noticeable improvement in IDD in mice after GCA treatment (Fig. 6E).
The ELISA test results showed that compared to the Sham group, the expression of the inflammatory factors IL-6, iNOS, TNF-α, and IL-1β in the serum of mice in the PBS group was significantly increased. However, after treatment, the expression of inflammatory factors in the serum of mice decreased, indicating a reduction in the inflammatory response in the intervertebral disc microenvironment. Furthermore, the inflammatory response in the intervertebral disc of mice was further reduced in the GCA treatment group compared to the GA treatment group (Fig. 6F). In addition, the TUNEL test results showed that treatment with GA and GCA reduced apoptosis of cells in the degenerated NP tissue, with GCA demonstrating a significantly stronger inhibitory effect on cell apoptosis compared to GA (Fig. 6G). Immunohistochemical staining results showed that treatment with GCA significantly decreased the expression of the CFIm25 protein in degenerated NP tissue of mice compared to the PBS or GA group (Fig. 6H). Furthermore, treatment with GCA significantly increased the expression of the COLII and Aggrecan proteins in degenerated NP tissue (Fig. 6I-J).
Based on these comprehensive assessments, we can conclude that the porous GelMA-HAMA hydrogel loaded with APET×2 polypeptide and sgRNA can effectively reduce the inflammatory response in the intervertebral disc immune microenvironment of mice, increase the expression of collagen protein (COLII) and aggrecan in the intervertebral disc microenvironment, alleviate the progression of IDD in mice, improve intervertebral disc bioproperties, and demonstrate potential for the treatment of IDD.
GCA alleviates inflammatory responses and IDD in mice by inhibiting the p38/NF-κB signaling pathway
To investigate the mechanisms of hydrogel-loaded polypeptide and sgRNA in the treatment of IDD in mice, we extracted proteins from the NP tissue of mice in the PBS treatment group and the GCA treatment group. Nanoparticle-based HPLC-MS/MS analysis, known as Data Independent Acquisition (DIA), was employed for quantitative proteomic analysis to explore the changes in protein content in the NP tissue of mice after GCA treatment (Figure S3).
DEPs were screened using the “limma” package in R language, with a criteria of p-value < 0.05 and |log2FC| > 1.0. A total of 194 DEPs, consisting of 73 upregulated and 121 downregulated proteins, were identified. The volcano plot and heat map showing the differentially expressed DEPs are presented in Fig. 7A-B. The 194 DEPs were subjected to GO functional enrichment analysis, which revealed their involvement in biological processes such as purine nucleotide metabolism, nucleotide metabolism, and ribose phosphate metabolism. In terms of cellular components, the DEPs were mainly enriched in extracellular matrix containing collagen, myelin sheath, and complexes containing mitochondrial proteins. Molecular function analysis showed enrichment in extracellular matrix structural constituents, magnesium ion binding, and catalytic activity associated with binding (Fig. 7C).

Proteomic sequencing analysis of critical signaling pathways mediating IDD. Note (A) Volcano plot showing differentially expressed genes in the IVD NP tissue of mice from the PBS treatment group (3 groups) and the GCA treatment group (3 groups). The yellow triangle represents upregulated genes, blue triangle represents downregulated genes, and black dots represent non-differentially expressed genes; (B) Heatmap of 194 DEPs, with white representing downregulation and blue representing upregulation; (C) Bubble plot of GO enrichment analysis for the 194 DEPs, with the color and size of circles representing the significance and number of enriched genes, respectively; (D) Bubble plot of KEGG enrichment analysis for the 194 DEPs, with the color and size of circles representing the significance and number of enriched genes, respectively; (E) Statistical analysis of PPI network interaction sites for the 194 DEPs, with a corresponding diagram; (F) PPI network graph of the 194 DEPs (Combined score = 0.7), with the color gradient from yellow to orange indicating the degree of the genes from small to large
Further enrichment analysis of the 122 overlapping genes was performed on KEGG pathways. The results showed that 76 overlapping genes were primarily involved in the NF-kappa B signaling pathway, mRNA surveillance pathway, TNF signaling pathway, Parkinson’s disease, and diabetic cardiomyopathy. Among these pathways, the NF-kappa B signaling pathway had the highest number of genes (Fig. 7D).
Additionally, protein-protein interaction network analysis using the STRING database revealed multiple potential interacting proteins around Rela and Mapk14. These proteins, encoded by p65 and p38, respectively, may serve as core regulators in the progression of IDD (Fig. 7E-F). According to literature reports, Nudt21 has been identified as an upstream regulatory factor in the NF-κB signaling pathway [55], while the APET×2 polypeptide has been shown to inhibit the p38 signaling pathway and attenuate the inflammatory response in IVD [35].
Therefore, we conducted RT-qPCR and Western blot analyses to examine the changes in p38 and p65 protein expression in degenerated NP cells following treatment with GA and GCA. The results revealed that compared to the Normal group, the Rela and Mapk14 mRNA expressions in the PBS group were significantly elevated, along with a significant increase in the phosphorylation levels of p38 and p65 proteins. In comparison to the PBS group, the GA treatment group exhibited a significant decrease in Mapk14 mRNA expression, while there was no notable change in Rela mRNA expression. However, the GCA treatment group showed a significant reduction in both Rela and Mapk14 mRNA expressions, along with a decrease in the phosphorylation levels of p38 protein in the GA treatment group and a significant decrease in the phosphorylation levels of both p38 and p65 proteins in the GCA treatment group. Additionally, the GCA treatment group exhibited a decrease in Rela and Mapk14 mRNA expressions and a decrease in the activation capacity of p38 and p65 phosphorylation levels, which was significantly stronger than the GA treatment group (Fig. 8A-B). RT-qPCR and Western blot analysis results also demonstrated that treatments with GA and GCA upregulated the expression of collagen COL II and aggregating proteoglycan Aggrecan, along with their encoding genes while downregulating the expression of NP degeneration-related factors COX-2 and MMP-3 proteins, and their encoding genes in degenerated NP cells. Moreover, the GCA treatment group exhibited a significantly stronger regulation effect on the respective proteins and their encoding genes when compared to the GA treatment group (Fig. 8C-D). Furthermore, immunohistochemical staining of mouse NP tissue slices after treatment revealed that in comparison to the PBS group or GA group, the phosphorylation levels of p38 and p65, as well as the protein expression of COX-2 and MMP-3, were significantly reduced in the intervertebral disc NP tissue of mice treated with GCA. This further confirmed the inhibitory effect of GCA on the p38/NF-κB signaling pathway (Fig. 8E-H).

GCA improves IDD through the p38/NF-κB signaling pathway. Note (A) RT-qPCR analysis of Mapk14 and Rela gene expression in NP cells from different treatment groups; (B) Western blot analysis of p38, p-p38, p65, and p-p65 protein expression in NP cells from different treatment groups; (C) RT-qPCR analysis of Col2a1, Acan, Mmp-3, and Ptgs2 gene expression in NP cells from different treatment groups; (D) Western blot analysis of COLII, Aggrecan, MMP-3, and COX-2 protein expression in NP cells from different treatment groups; (E) Immunohistochemistry staining showing the expression of p-p38 protein in mouse intervertebral disc NP tissue (scale bar in the legend = 200 μm); (F) Immunohistochemistry staining showing the expression of p-p65 protein in mouse intervertebral disc NP tissue (scale bar in the legend = 200 μm); (G) Immunohistochemistry staining showing the expression of MMP-3 protein in mouse intervertebral disc NP tissue (scale bar in the legend = 200 μm); (H) Immunohistochemistry staining showing the expression of COX-2 protein in mouse intervertebral disc NP tissue (scale bar in the legend = 200 μm). The quantitative data shown in the figure are presented as Mean ± SD, with cell experiments repeated 3 times per group and animal experiments conducted with 6 mice per group. * indicates comparison between the two groups, with significance levels as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
The above findings confirm that GCA alleviates inflammation reactions in the degenerated intervertebral disc immune microenvironment by inhibiting the p38/NF-κB signaling pathway, thus improving IDD in mice.
GCA regulates the immune microenvironment of the intervertebral disc to promote NP regeneration
According to the literature, immune cells, including B cells and T cells, are believed to secrete molecules that promote inflammation, autophagy, or cell apoptosis (such as TNF-α and IL-1β), leading to IDD. Additionally, immune cells release a significant amount of inflammatory cytokines, exacerbating the cascade of inflammatory reactions [56, 57]. Given the aforementioned single-cell sequencing results indicating that immune cells play a crucial role in mediating cell-to-cell communication in IDD, we further investigated the impact of GCA on immune cells in the IDD immune microenvironment.
Flow cytometry analysis revealed that, compared to the Sham group, the number of T cells and B cells in the NP tissue of mice in the PBS group significantly increased, promoting intervertebral disc inflammation. In contrast, compared to the PBS group, the numbers of T cells and B cells in the NP tissue of mice in the GA and GCA groups significantly decreased. Notably, the reduction in immune cell numbers in the GCA group was significantly greater than that in the GA group (Fig. 9A). Furthermore, as T cells were found to be more abundant than B cells, we further analyzed the changes in the numbers of CD4+ and CD8+ T cells within the T cell population. The results revealed a significant increase in the numbers of CD4+ and CD8+ T cells in the NP tissue of mice with IDD. However, following treatment with either GA or GCA, the cell numbers significantly decreased. Interestingly, the treatment effectiveness of GCA was greater than that of GA (Fig. 9B).

GCA regulation improves immune microenvironment in IDD. Note (A) Changes in the number of T cells and B cells in the NP tissue of mice in different treatment groups; (B) Changes in the number of CD4+ T cells and CD8+ T cells in the NP tissue of mice in different treatment groups; (C) Changes in the number of Th1 cells in the NP tissue of mice in different treatment groups; (D) Changes in the number of Th2 cells in the NP tissue of mice in different treatment groups; (E) Expression changes of cell factors IFN-γ, IL-12, IL-6, and IL-13 in NP tissue of mice in different treatment groups detected by Western blot. Quantitative data shown in the figure are presented as Mean ± SD, with 6 mice included in each animal experiment. * indicates comparison between the two groups, with significance levels as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Furthermore, we observed that the number of CD4+ T cells exceeded that of CD8+ T cells. Research indicates that T cell activation serves as a unique signal for cell-mediated autoimmunity, with Th1 and Th2 cells playing fundamental roles in the immune regulation of IDD. Th1 cells primarily produce pro-inflammatory cytokines, such as IFN-γ and IL-12, contributing to inflammation and cell-mediated immune responses, while Th2 cells produce anti-inflammatory cytokines like IL-6 and IL-13, participating in humoral immune responses [54, 57]. Subsequently, we further investigated the changes in the numbers of Th1 and Th2 cells within CD4+ T cells. Our analysis revealed that, compared to the Sham group, the number of Th1 cells in the NP tissue of mice in the PBS group significantly increased, while the number of Th2 cells decreased significantly. Following treatment with GA and GCA, the number of Th1 cells decreased while the number of Th2 cells increased in the NP tissue of mice. Importantly, the treatment effect of GCA was significantly stronger than that of GA (Fig. 9C-D).
Furthermore, we conducted Western blot analysis to validate that GCA treatment can decrease the number of Th1 cells and increase the number of Th2 cells. The results revealed (Fig. 9E) that, compared to the Sham group, the expression of the pro-inflammatory cytokines IFN-γ and IL-12 increased in the NP tissue of mice in the PBS group, while the expression of anti-inflammatory cytokines IL-6 and IL-13 decreased. Following treatment with GA and GCA, the expression of IFN-γ and IL-12 significantly decreased, while the expression of IL-6 and IL-13 significantly increased. Importantly, GCA exhibited a significantly stronger inhibitory effect on immune inflammation in the intervertebral disc of mice than GA.
These research findings demonstrate that GCA can modulate the immune microenvironment in the NP tissue of IDD mice, thereby promoting the regeneration of degenerated intervertebral disc NP.
Discussion
IDD is a degenerative disease associated with various factors such as age, genetics, and mechanical load, and is considered a significant cause of morbidity and mortality in clinical practice [58, 59]. IDD can lead to lumbar back pain and other spinal-related conditions, profoundly affecting patients’ quality of life. Although the exact pathological mechanisms remain incompletely understood, research indicates that cytokines, growth factors, enzymes, and other proteins play crucial roles in the process of IDD [60]. Therefore, in-depth exploration of these molecules and their roles in IDD holds substantial importance for developing new therapeutic strategies.
This study focused on CFIm25 as a target and investigated the mechanism of NP regeneration in degenerated IVDs by utilizing APET×2 peptide and sgRNA loaded in GelMA-HAMA hydrogels. Previous studies have highlighted that IDD is the result of the combined effects of multiple factors [17, 61, 62]. Some studies suggest that inflammatory reactions play a crucial role in the onset and progression of IDD, while others emphasize the significance of cell proliferation and migration [63, 64]. This study delved into the expression changes of CFIm25 in IDD and its role in NP cell function, affirming the importance of CFIm25 in the pathogenesis of IDD [10, 17, 65, 66].
Cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) are the major pro-inflammatory cytokines with potent inflammatory activities, capable of promoting the secretion of various pro-inflammatory mediators. In degenerative IDD, their levels are upregulated and closely associated with various pathological processes of IDD, including inflammatory reactions, matrix destruction, cellular senescence, autophagy, apoptosis, pyroptosis, and proliferation, being considered as crucial mediators contributing to IDD [67]. In this study, we extensively investigated how the expression inhibition of CFIm25 could slow down IDD by enhancing the immune microenvironment. Our findings indicate that the strategy of loading APET×2 peptide and sgRNA for CFIm25 knockout into GelMA-HAMA hydrogels significantly reduced the expression of inflammatory factors IL-6, iNOS, IL-1β, and TNF-α in the IVD microenvironment, while also increasing the expression of collagen COLII and aggregating proteoglycan Aggrecan. These discoveries underscore the crucial role of the immune microenvironment in the development of IDD, providing novel therapeutic targets for future research.
In terms of methodology, this study utilized transcriptomics and high-throughput transcriptomics analysis to identify factors significantly associated with IDD, which were further validated through RT-qPCR and Western blot [68]. Compared to other studies, we employed CRISPR-Cas9 technology to knockout CFIm25 and overexpressed CFIm25 using lentiviral vectors to establish NP cell lines with CFIm25 knockout and overexpression [69,70,71]. These methods provide more precise tools for research and a more detailed understanding of the role of CFIm25 in IDD [72, 73].
In terms of experimental results, we observed that inhibiting CFIm25 improved the degeneration of NP cells and promoted their proliferation and migration capacity. These results are consistent with other research, highlighting the important role of CFIm25 in IDD [74, 75]. Additionally, we successfully prepared GelMA-HAMA hydrogel and loaded the peptide APET×2 and sgRNA onto it, demonstrating its effectiveness in promoting NP regeneration in vitro and in a mouse model. These findings provide new therapeutic strategies for the treatment of IDD.
This study presents several differences compared to previous studies. Traditional treatment methods mainly focus on pain relief and improving quality of life without addressing the restoration of intervertebral disc tissue function [76,77,78]. However, this study proposes a new treatment strategy by improving the function of NP cells through targeted regulation of CFIm25 and evaluating its effectiveness using the GelMA-HAMA hydrogel both in vitro and in vivo [79]. This approach provides a new possibility for intervertebral disc regeneration.
In summary, this study first discovered through multi-omics analyses that the CFIm25 encoding gene, Nudt21, plays an essential role in the progression of intervertebral disc degenerative diseases, with increased expression in degenerated discs. Moreover, in vitro intervention experiments targeting Nudt21 further confirmed that knocking down CFIm25 expression can improve IDD in mice. Based on these findings, this study successfully developed a novel porous spherical GelMA-HAMA hydrogel as a carrier, loaded with an APET×2 polypeptide and sgRNA to specifically target CFIm25, aiming to alleviate the symptoms of IDD. Our strategy breaks new ground by targeting CFIm25 to regulate the intervertebral disc immune microenvironment and promote the regeneration of degenerated intervertebral disc NP. In vitro and in vivo experiments, as well as proteomic analysis confirmed that the novel hydrogel GCA can reduce CFIm25 protein expression, inhibit the expression of inflammatory factors IL-6, iNOS, IL-1β, and TNF-α in the intervertebral disc immune microenvironment by suppressing the p38/NF-κB signaling pathway, reduce inflammation in the intervertebral disc microenvironment, increase the expression of collagen protein COLII and aggregating protein aggrecan, suppress the expression of degeneration-related factors COX-2 and MMP-3 in NP, and thereby improve IDD in mice.
The scientific and clinical value of this study lies in its in-depth exploration of the pathogenesis of IDD and the proposal of a new treatment strategy. By targeting CFIm25 in NP cells and utilizing the porous spherical GelMA-HAMA hydrogel loaded with APET×2 polypeptide and sgRNA, researchers successfully promoted the regeneration of degenerated intervertebral disc NP and improved the progression of IDD. This study is of significant importance for the diagnosis and treatment of IDD patients, providing valuable guidance for the search for new therapeutic approaches.
However, this study has several limitations. Firstly, the experimental subjects were limited to mouse and in vitro models, lacking human validation due to species differences and physiological complexities. Therefore, the direct applicability of these findings to the clinical environment is questionable, and their feasibility and safety in clinical applications remain to be verified. Secondly, although the study results indicate that targeting and regulating CFIm25 and utilizing APETx2 peptide and sgRNA can improve IDD, the specific mechanism has not been fully elucidated. Further research is still needed to delve into the treatment effects and mechanisms.
Looking ahead, this study provides many intriguing directions for future research. Firstly, a more detailed understanding of the role of CFIm25 in IDD. Secondly, considering the combination of this treatment method with other therapies could explore more effective treatment strategies. Additionally, the potential application of the porous spherical GelMA-HAMA hydrogel in the treatment of other degenerative diseases could also be further studied. These further investigations will contribute to a deeper understanding of IDD treatment and provide better treatment choices for patients. In future research, it is crucial to validate the expression levels of CFIm25 in human clinical samples to further confirm its role as a potential therapeutic target for degenerative intervertebral disc disease. This step will help bridge the gap between preclinical research findings and clinical applications, thus providing stronger support for the development of effective treatment strategies.
Data availability
No datasets were generated or analysed during the current study.
References
Fine N, Lively S, Séguin CA, Perruccio AV, Kapoor M, Rampersaud R. Intervertebral disc degeneration and osteoarthritis: a common molecular disease spectrum. Nat Rev Rheumatol. 2023;19(3):136–52. https://doi.org/10.1038/s41584-022-00888-z.
Zhang GZ, Liu MQ, Chen HW, et al. NF-κB signalling pathways in nucleus pulposus cell function and intervertebral disc degeneration. Cell Prolif. 2021;54(7):e13057. https://doi.org/10.1111/cpr.13057.
Wang Y, Cheng H, Wang T, Zhang K, Zhang Y, Kang X. Oxidative stress in intervertebral disc degeneration: molecular mechanisms, pathogenesis and treatment. Cell Prolif. 2023;56(9):e13448. https://doi.org/10.1111/cpr.13448.
Yabe Y, Hagiwara Y, Tsuchiya M, et al. Factors Associated with Thickening of the Ligamentum Flavum on Magnetic Resonance Imaging in patients with lumbar spinal canal stenosis. Spine (Phila Pa 1976). 2022;47(14):1036–41. https://doi.org/10.1097/BRS.0000000000004341.
Yang XX, Yip CH, Zhao S, Ho YP, Chan BP. A bio-inspired nano-material recapitulating the composition, ultra-structure, and function of the glycosaminoglycan-rich extracellular matrix of nucleus pulposus. Biomaterials. 2023;293:121991. https://doi.org/10.1016/j.biomaterials.2022.121991.
Liu Z, Wang H, Yuan Z et al. High-resolution 3D printing of angle-ply annulus fibrosus scaffolds for intervertebral disc regeneration. Biofabrication. 2022;15(1):https://doi.org/10.1088/1758-5090/aca71f. Published 2022 Dec 15. doi:10.1088/1758-5090/aca71f.
Qian H, He L, Ye Z, Wei Z, Ao J. Decellularized matrix for repairing intervertebral disc degeneration: fabrication methods, applications and animal models. Mater Today Bio. 2022;18:100523. https://doi.org/10.1016/j.mtbio.2022.100523. Published 2022 Dec 17.
Kawabata K, Akeda S, Yamada J, et al. Advances in Platelet-Rich plasma treatment for spinal diseases: a systematic review. Int J Mol Sci. 2023;24(8):7677. https://doi.org/10.3390/ijms24087677. Published 2023 Apr 21.
Lindsey T, Dydyk AM. Spinal osteoarthritis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; July 9, 2023.
Ou-Yang DC, Kleck CJ, Ackert-Bicknell CL. Genetics of Intervertebral Disc Degeneration. Curr Osteoporos Rep. 2023;21(1):56–64. https://doi.org/10.1007/s11914-022-00769-0.
Zhang Y, Han S, Kong M, Tu Q, Zhang L, Ma X. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthritis Cartilage. 2021;29(9):1324–34. https://doi.org/10.1016/j.joca.2021.06.010.
Yu P, Mao F, Chen J, et al. Characteristics and mechanisms of resorption in lumbar disc herniation. Arthritis Res Ther. 2022;24(1):205. https://doi.org/10.1186/s13075-022-02894-8. Published 2022 Aug 23.
Sun K, Jiang J, Wang Y, et al. The role of nerve fibers and their neurotransmitters in regulating intervertebral disc degeneration. Ageing Res Rev. 2022;81:101733. https://doi.org/10.1016/j.arr.2022.101733.
Wang L, He T, Liu J, et al. Revealing the Immune Infiltration Landscape and identifying diagnostic biomarkers for lumbar disc herniation. Front Immunol. 2021;12:666355. https://doi.org/10.3389/fimmu.2021.666355. Published 2021 May 27.
Zhang J, Sun J, Chen D, et al. Suppression of matrix degradation and amelioration of disc degeneration by a 970-nm diode laser via inhibition of the p38 MAPK pathway in a rabbit model. Lasers Med Sci. 2023;38(1):58. https://doi.org/10.1007/s10103-023-03717-1. Published 2023 Jan 31.
Li X, Wang X, Chen C, et al. Accumulation of NCOA1 dependent on HERC3 deficiency transactivates matrix metallopeptidases and promotes extracellular matrix degradation in intervertebral disc degeneration. Life Sci. 2023;320:121555. https://doi.org/10.1016/j.lfs.2023.121555.
Xin J, Wang Y, Zheng Z, Wang S, Na S, Zhang S. Treatment of intervertebral disc degeneration. Orthop Surg. 2022;14(7):1271–80. https://doi.org/10.1111/os.13254.
Chen S, Lei L, Li Z, et al. Grem1 accelerates nucleus pulposus cell apoptosis and intervertebral disc degeneration by inhibiting TGF-β-mediated Smad2/3 phosphorylation. Exp Mol Med. 2022;54(4):518–30. https://doi.org/10.1038/s12276-022-00753-9.
Bhujel B, Shin HE, Choi DJ, Han I. Mesenchymal Stem Cell-Derived Exosomes and Intervertebral Disc Regeneration: Review. Int J Mol Sci. 2022;23(13):7306. Published 2022 Jun 30. https://doi.org/10.3390/ijms23137306
Brisby H. Pathology and possible mechanisms of nervous system response to disc degeneration. J Bone Joint Surg Am. 2006;88(Suppl 2):68–71. https://doi.org/10.2106/JBJS.E.01282.
Vergroesen PP, Kingma I, Emanuel KS, et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthritis Cartilage. 2015;23(7):1057–70. https://doi.org/10.1016/j.joca.2015.03.028.
Vo NV, Hartman RA, Yurube T, Jacobs LJ, Sowa GA, Kang JD. Expression and regulation of metalloproteinases and their inhibitors in intervertebral disc aging and degeneration. Spine J. 2013;13(3):331–41. https://doi.org/10.1016/j.spinee.2012.02.027.
Ali F, Khan I, Chen J, Akhtar K, Bakhsh EM, Khan SB. Emerging Fabrication Strategies of Hydrogels and Its Applications. Gels. 2022;8(4):205. Published 2022 Mar 24. https://doi.org/10.3390/gels8040205
Du L, Yang Q, Zhang J, et al. Engineering a biomimetic integrated scaffold for intervertebral disc replacement. Mater Sci Eng C Mater Biol Appl. 2019;96:522–9. https://doi.org/10.1016/j.msec.2018.11.087.
Chen Z, Liu F, Chen Y, et al. Targeted delivery of CRISPR/Cas9-Mediated Cancer Gene Therapy via Liposome-Templated Hydrogel Nanoparticles. Adv Funct Mater. 2017;27(46):1703036. https://doi.org/10.1002/adfm.201703036.
Kalisky T, Blainey P, Quake SR. Genomic analysis at the single-cell level. Annu Rev Genet. 2011;45:431–45. https://doi.org/10.1146/annurev-genet-102209-163607.
Wang T, Wang L, Zhang L, Long Y, Zhang Y, Hou Z. Single-cell RNA sequencing in orthopedic research. Bone Res. 2023;11(1):10. https://doi.org/10.1038/s41413-023-00245-0. Published 2023 Feb 24.
Pozojevic J, Spielmann M. Single-cell sequencing in neurodegenerative disorders. Mol Diagn Ther. 2023;27(5):553–61. https://doi.org/10.1007/s40291-023-00668-9.
Chang X, Zheng Y, Xu K, Single-Cell RNA, Sequencing. Technological Progress and Biomedical Application in Cancer Research. Mol Biotechnol. 2024;66(7):1497–519. https://doi.org/10.1007/s12033-023-00777-0.
Gupta RC, Lall R, Srivastava A, Sinha A. Hyaluronic Acid: Molecular mechanisms and Therapeutic Trajectory. Front Vet Sci. 2019;6:192. https://doi.org/10.3389/fvets.2019.00192. Published 2019 Jun 25.
Schuurman W, Levett PA, Pot MW, et al. Gelatin-methacrylamide hydrogels as potential biomaterials for fabrication of tissue-engineered cartilage constructs. Macromol Biosci. 2013;13(5):551–61. https://doi.org/10.1002/mabi.201200471.
Eke G, Mangir N, Hasirci N, MacNeil S, Hasirci V. Development of a UV crosslinked biodegradable hydrogel containing adipose derived stem cells to promote vascularization for skin wounds and tissue engineering. Biomaterials. 2017;129:188–98. https://doi.org/10.1016/j.biomaterials.2017.03.021.
Rohanifar M, Clayton SW, Easson GWD, et al. Single cell RNA-Sequence analyses reveal uniquely expressed genes and heterogeneous Immune cell involvement in the rat model of intervertebral disc degeneration. Appl Sci (Basel). 2022;12(16):8244. https://doi.org/10.3390/app12168244.
Che H, Li J, Li Y et al. p16 deficiency attenuates intervertebral disc degeneration by adjusting oxidative stress and nucleus pulposus cell cycle. Elife. 2020;9:e52570. Published 2020 Mar 3. https://doi.org/10.7554/eLife.52570
Bian J, Cai F, Chen H, et al. Modulation of local overactive inflammation via Injectable Hydrogel Microspheres. Nano Lett. 2021;21(6):2690–8. https://doi.org/10.1021/acs.nanolett.0c04713.
Meng Q, Liu K, Liu Z, et al. Digoxin protects against intervertebral disc degeneration via TNF/NF-κB and LRP4 signaling. Front Immunol. 2023;14:1251517. https://doi.org/10.3389/fimmu.2023.1251517. Published 2023 Sep 18.
Tam V, Chan WCW, Leung VYL, et al. Histological and reference system for the analysis of mouse intervertebral disc. J Orthop Res. 2018;36(1):233–43. https://doi.org/10.1002/jor.23637.
Han S, Zhang Y, Zhang X, et al. Single-cell RNA sequencing of the Nucleus Pulposus reveals chondrocyte differentiation and regulation in intervertebral disc degeneration. Front Cell Dev Biol. 2022;10:824771. https://doi.org/10.3389/fcell.2022.824771. Published 2022 Feb 21.
Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. https://doi.org/10.1093/nar/gkv007.
Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–e358729. https://doi.org/10.1016/j.cell.2021.04.048.
Tsingas M, Ottone OK, Haseeb A, et al. Sox9 deletion causes severe intervertebral disc degeneration characterized by apoptosis, matrix remodeling, and compartment-specific transcriptomic changes. Matrix Biol. 2020;94:110–33. https://doi.org/10.1016/j.matbio.2020.09.003.
Ayturk U. RNA-seq in skeletal Biology. Curr Osteoporos Rep. 2019;17(4):178–85. https://doi.org/10.1007/s11914-019-00517-x.
Simoneau J, Dumontier S, Gosselin R, Scott MS. Current RNA-seq methodology reporting limits reproducibility. Brief Bioinform. 2021;22(1):140–5. https://doi.org/10.1093/bib/bbz124.
Bratsman A, Couasnay G, Elefteriou F. A step-by-step protocol for isolation of murine nucleus pulposus cells. JOR Spine. 2019;2(4):e1073. https://doi.org/10.1002/jsp2.1073. Published 2019 Dec 19.
Zheng B, Li S, Xiang Y, et al. Netrin-1 mediates nerve innervation and angiogenesis leading to discogenic pain. J Orthop Translat. 2022;39:21–33. https://doi.org/10.1016/j.jot.2022.11.006. Published 2022 Dec 23.
Shao Z, Wang B, Shi Y, et al. Senolytic agent Quercetin ameliorates intervertebral disc degeneration via the Nrf2/NF-κB axis. Osteoarthritis Cartilage. 2021;29(3):413–22. https://doi.org/10.1016/j.joca.2020.11.006.
Xu H, Wei K, Tu J, et al. Reducing inflammation and Vascular Invasion in Intervertebral Disc Degeneration via Cystathionine-γ-Lyase inhibitory effect on E-Selectin. Front Cell Dev Biol. 2021;9:741046. https://doi.org/10.3389/fcell.2021.741046. Published 2021 Nov 15.
Zhang Z, Zhang L, Yang J, et al. Influence of extracellular nanovesicles derived from adipose-derived stem cells on nucleus pulposus cell from patients with intervertebral disc degeneration. Exp Ther Med. 2021;22(6):1431. https://doi.org/10.3892/etm.2021.10866.
Tellier M, Hardy JG, Norbury CJ, Murphy S. Effect of CFIm25 knockout on RNA polymerase II transcription. BMC Res Notes. 2018;11(1):894. https://doi.org/10.1186/s13104-018-4006-1. Published 2018 Dec 14.
Yun Z, Wang Y, Feng W, Zang J, Zhang D, Gao Y. Overexpression of microRNA-185 alleviates intervertebral disc degeneration through inactivation of the Wnt/β-catenin signaling pathway and downregulation of Galectin-3. Mol Pain. 2020;16:1744806920902559. https://doi.org/10.1177/1744806920902559.
Xia X, Liu Y, Lu Y, et al. Retuning mitochondrial Apoptosis/Mitophagy balance via SIRT3-Energized and Microenvironment-Modulated Hydrogel microspheres to impede osteoarthritis. Adv Healthc Mater. 2023;12(32):e2302475. https://doi.org/10.1002/adhm.202302475.
Zhu D, Li Z, Huang K, Caranasos TG, Rossi JS, Cheng K. Minimally invasive delivery of therapeutic agents by hydrogel injection into the pericardial cavity for cardiac repair. Nat Commun. 2021;12(1):1412. Published 2021 Mar 3. https://doi.org/10.1038/s41467-021-21682-7
Kudelko M, Chen P, Tam V, et al. PRIMUS: Comprehensive proteomics of mouse intervertebral discs that inform novel biology and relevance to human disease modelling. Matrix Biol Plus. 2021;12:100082. https://doi.org/10.1016/j.mbplus.2021.100082. Published 2021 Jul 24.
Yao Y, Xue H, Chen X, et al. Polarization of helper T lymphocytes maybe involved in the pathogenesis of lumbar disc herniation. Iran J Allergy Asthma Immunol. 2017;16(4):347–57.
Lou JC, Lan YL, Gao JX, et al. Silencing NUDT21 attenuates the mesenchymal identity of Glioblastoma Cells via the NF-κB pathway. Front Mol Neurosci. 2017;10:420. https://doi.org/10.3389/fnmol.2017.00420. Published 2017 Dec 19.
Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10(1):44–56. https://doi.org/10.1038/nrrheum.2013.160.
Hai B, Song Q, Du C, et al. Comprehensive bioinformatics analyses reveal immune genes responsible for altered immune microenvironment in intervertebral disc degeneration. Mol Genet Genomics. 2022;297(5):1229–42. https://doi.org/10.1007/s00438-022-01912-3.
Adams MA, Roughley PJ. What is intervertebral disc degeneration, and what causes it? Spine (Phila Pa 1976). 2006;31(18):2151–61. https://doi.org/10.1097/01.brs.0000231761.73859.2c.
Kos N, Gradisnik L, Velnar T. A brief review of the degenerative intervertebral disc disease. Med Arch. 2019;73(6):421–4. https://doi.org/10.5455/medarh.2019.73.421-424.
Cherif H, Mannarino M, Pacis AS, et al. Single-cell RNA-Seq analysis of cells from degenerating and non-degenerating intervertebral discs from the same individual reveals new biomarkers for intervertebral disc degeneration. Int J Mol Sci. 2022;23(7):3993. https://doi.org/10.3390/ijms23073993. Published 2022 Apr 3.
Jin P, Xing Y, Xiao B, et al. Diabetes and intervertebral disc degeneration: a mendelian randomization study. Front Endocrinol (Lausanne). 2023;14:1100874. https://doi.org/10.3389/fendo.2023.1100874. Published 2023 Feb 28.
Wang W, Jing X, Du T, et al. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic Biol Med. 2022;190:234–46. https://doi.org/10.1016/j.freeradbiomed.2022.08.018.
Zheng Y, Chen X, Lan T, Yan B, Zhang R. Panax notoginseng saponin reduces IL-1β-stimulated apoptosis and endoplasmic reticulum stress of nucleus pulposus cells by suppressing miR-222-3p. Ann Transl Med. 2022;10(13):748. https://doi.org/10.21037/atm-22-3203.
Li T, Peng Y, Chen Y, et al. Long intergenic non-coding RNA – 00917 regulates the proliferation, inflammation, and pyroptosis of nucleus pulposus cells via targeting miR-149-5p/NOD-like receptor protein 1 axis. Bioengineered. 2022;13(3):6036–47. https://doi.org/10.1080/21655979.2022.2043100.
Cazzanelli P, Wuertz-Kozak K. MicroRNAs in intervertebral disc degeneration, apoptosis, inflammation, and mechanobiology. Int J Mol Sci. 2020;21(10):3601. https://doi.org/10.3390/ijms21103601. Published 2020 May 20.
Koroth J, Buko EO, Abbott R et al. Macrophages and Intervertebral Disc Degeneration. Int J Mol Sci. 2023;24(2):1367. Published 2023 Jan 10. https://doi.org/10.3390/ijms24021367
Wang Y, Che M, Xin J, Zheng Z, Li J, Zhang S. The role of IL-1β and TNF-α in intervertebral disc degeneration. Biomed Pharmacother. 2020;131:110660. https://doi.org/10.1016/j.biopha.2020.110660.
Zhang C, Feng YG, Tam C, Wang N, Feng Y. Transcriptional profiling and machine learning unveil a concordant biosignature of type I Interferon-Inducible Host Response across Nasal Swab and pulmonary tissue for COVID-19 diagnosis. Front Immunol. 2021;12:733171. https://doi.org/10.3389/fimmu.2021.733171. Published 2021 Nov 22.
Endo F, Kasai A, Soto JS, et al. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science. 2022;378(6619):eadc9020. https://doi.org/10.1126/science.adc9020.
Clark IC, Wheeler MA, Lee HG, et al. Identification of astrocyte regulators by nucleic acid cytometry. Nature. 2023;614(7947):326–33. https://doi.org/10.1038/s41586-022-05613-0.
Costamagna G, Comi GP, Corti S. Advancing Drug Discovery for neurological disorders using iPSC-Derived neural organoids. Int J Mol Sci. 2021;22(5):2659. https://doi.org/10.3390/ijms22052659. Published 2021 Mar 6.
Romanens L, Chaskar P, Marcone R, et al. Clonal expansion of intra-epithelial T cells in breast cancer revealed by spatial transcriptomics. Int J Cancer. 2023;153(9):1568–78. https://doi.org/10.1002/ijc.34620.
Zheng Y, Yang X. Application and prospect of single-cell sequencing in cancer metastasis. Future Oncol. 2022;18(21):2723–36. https://doi.org/10.2217/fon-2022-0156.
Masamha CP. The emerging roles of CFIm25 (NUDT21/CPSF5) in human biology and disease. Wiley Interdiscip Rev RNA. 2023;14(3):e1757. https://doi.org/10.1002/wrna.1757.
Ran Y, Huang S, Shi J, et al. CFIm25 regulates human stem cell function independently of its role in mRNA alternative polyadenylation. RNA Biol. 2022;19(1):686–702. https://doi.org/10.1080/15476286.2022.2071025.
Olby NJ, Moore SA, Brisson B, et al. ACVIM consensus statement on diagnosis and management of acute canine thoracolumbar intervertebral disc extrusion. J Vet Intern Med. 2022;36(5):1570–96. https://doi.org/10.1111/jvim.16480.
Krut Z, Pelled G, Gazit D, Gazit Z. Stem cells and exosomes: New therapies for intervertebral disc degeneration. Cells. 2021;10(9):2241. https://doi.org/10.3390/cells10092241. Published 2021 Aug 29.
Tu J, Li W, Yang S, et al. Single-cell transcriptome profiling reveals multicellular ecosystem of Nucleus Pulposus during Degeneration Progression. Adv Sci (Weinh). 2022;9(3):e2103631. https://doi.org/10.1002/advs.202103631.
Cai Y, Chen Z, Liang Y, et al. Cleavage factor Im 25 as a potential biomarker for prognosis of colorectal cancer. Transl Cancer Res. 2021;10(12):5267–79. https://doi.org/10.21037/tcr-21-1441.
Acknowledgements
Not applicable.
Funding
This study was supported by National Natural Science Foundation of China Key Program (Project Approval No. 81830077).
Author information
Authors and Affiliations
Contributions
Xiao-Jun Yu and Yuan-Ting Zhao: Conceptualization, methodology, data curation, and writing—original draft preparation. Haimiti Abudouaini and Peng Zou: Validation, formal analysis, and investigation. Tian-Qi Li and Xiao-Fan Bai: Data curation and resources. Shan-Xi Wang and Jian-Bin Guan: Supervision, project administration, and funding acquisition. Meng-wei Li: Software, visualization, and writing—review and editing. Xiao-dong Wang: Revision, Software, visualization, and writing—review and editing. Ying-guang Wang and Ding-Jun Hao: Supervision, project administration, funding acquisition, and correspondence. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
All experimental protocols have been approved by Honghui Hospital, Xi’an Jiaotong University’s ethical review committee. We have taken great care to ensure the welfare of the mice involved in the experiment.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Yu, XJ., Zhao, YT., Abudouaini, H. et al. A novel spherical GelMA-HAMA hydrogel encapsulating APET×2 polypeptide and CFIm25-targeting sgRNA for immune microenvironment modulation and nucleus pulposus regeneration in intervertebral discs. J Nanobiotechnol 22, 556 (2024). https://doi.org/10.1186/s12951-024-02783-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-024-02783-z