
Abstract
Post-translational SUMOylation of nuclear and cytosolic proteins maintains homeostasis in eukaryotic cells and orchestrates programmed responses to changes in metabolic demand or extracellular stimuli. In excitable cells, SUMOylation tunes the biophysical properties and trafficking of ion channels. Ion channel SUMOylation status is determined by the opposing enzyme activities of SUMO ligases and deconjugases. Phosphorylation also plays a permissive role in SUMOylation. SUMO deconjugases have been identified for several ion channels, but their corresponding E3 ligases remain unknown. This study shows PIAS3, a.k.a. KChAP, is a bona fide SUMO E3 ligase for Kv4.2 and HCN2 channels in HEK cells, and endogenous Kv4.2 and Kv4.3 channels in cardiomyocytes. PIAS3-mediated SUMOylation at Kv4.2-K579 increases channel surface expression through a rab11a-dependent recycling mechanism. PKA phosphorylation at Kv4.2-S552 reduces the current mediated by Kv4 channels in HEK293 cells, cardiomyocytes, and neurons. This study shows PKA mediated phosphorylation blocks Kv4.2-K579 SUMOylation in HEK cells and cardiomyocytes. Together, these data identify PIAS3 as a key downstream mediator in signaling cascades that control ion channel surface expression.
Similar content being viewed by others

Background
Post-translational SUMOylation fine-tunes the functional expression of ion channels [1, 2], thereby shaping the intrinsic properties of excitable cells [3,4,5,6,7,8]. SUMOylation levels are dynamically adjusted by re-balancing the activities of several enzymes [9, 10]. SUMO peptides are conjugated to lysine (K) residues on target proteins by the single conjugating enzyme, ubc9. The majority of SUMOylation (~ 65%) occurs within identifiable consensus sequences [11]. Under certain conditions, ubc9 can by itself effect SUMOylation at a consensus sequence, but more often, it cooperates with a SUMO E3 ligase [12]. A SUMO E3 ligase has two functions: it links ubc9 to the target protein, and it optimizes the orientation of the donor SUMO loaded onto ubc9 so that it can be efficiently conjugated to the K on the target protein. SUMO E3 ligase activity is opposed by SUMO deconjugases, like Sentrin-specific proteases (SENPs), that remove SUMO from target proteins [13]. Ion channel regulation by SENPs has been investigated in multiple cell types [5,6,7, 14, 15]. In contrast, no bona fide SUMO E3 ligase has been identified for any ion channel to date.
Protein inhibitor of activated stat proteins (PIAS1-4) belong to the siz/PIAS (SP)-really interesting gene (RING) family of SUMO E3 ligases. All PIAS proteins contain a SP-RING domain and two SUMO interaction motifs (SIM1 & SIM2) [16,17,18,19]. The SP-RING domain binds a charged ubc9 (loaded with a donor SUMO) and locks it in an active conformation. The SIM domains bind the donor SUMO and a second SUMO conjugated to the backside of ubc9; this properly orients the enzyme for conjugation of the donor SUMO to the target. All three domains are thought to be necessary for PIAS catalytic activity. Decades-old investigations suggest that PIAS3 may be a SUMO E3 ligase for several ion channels. The protein, KChAP, was initially identified in a yeast 2-hybrid screen using a rat heart library and a voltage-gated potassium (Kv) channel β-subunit (Kvβ1) as bait [20]. KChAP not only interacted with Kvβ1 and Kvβ2, but also with Kv α-subunits including Kv1 isoforms (Kv1.1-5), Kv2 isoforms (Kv2.1/2), and the Kv4.3 channel [20,21,22]. KChAP had diverse effects when co-expressed with ion channels in heterologous systems. It altered currents mediated by several Kv α-subunits and prevented interactions between some Kv α- and β-subunits. The authors eventually realized that KChAP was identical to PIAS3, which at that time had not yet been identified as a SUMO E3 ligase. Studies on KChAP terminated before that discovery was made in 2001 [23, 24]. As a result, the idea that PIAS3 is a SUMO E3 ligase for cardiomyocyte Kv channels has never been tested; however, it is noteworthy that PIAS3 is known to associate with the SUMO substrate, GluR6a, in neurons [25].
Kv4 channels mediate a fast, transient outward current called IA in neurons and Itof in cardiomyocytes. The SUMOylation sites on Kv4 α-subunits are conserved across isoforms and species [26]. Increasing KChAP and increasing SUMOylation seem to produce like effects on Kv4 channels. Early studies using heterologous co-expression in Xenopus oocytes and mouse L-cells showed that KChAP co-expression significantly increased Kv4.3 channel surface expression relative to Kv4.3 alone [22]. More recent work showed that enhanced SUMOylation at Kv4.2-K579 increased channel surface expression by augmenting rab11-dependent recycling of endocytosed channels [27, 28]. The parallel results from the two lines of study suggest that PIAS3 could be a SUMO E3 ligase for Kv4 channels, but there is one piece of the puzzle that does not fit this hypothesis. Early work on KChAP identified a 98 amino acid fragment of the KChAP protein, termed the M-fragment, that could not only bind to Kv α- and β-subunits, but could also increase α-subunit surface expression [21, 22]. The M-fragment lacked the SIM2 domain and most of the SP-RING domain but contained the complete SIM1 domain. As previously stated, SIM1 is necessary for PIAS3 catalytic activity, but it was never shown to be sufficient [18, 19]. Since SUMO E3 ligases can have SUMO-independent functions, sometimes mediated by the SIM1 domain [29,30,31], it is necessary to provide compelling data to show that PIAS3 is a bona fide SUMO E3 ligase for ion channels. Here we used heterologous expression studies in HEK cells and investigations on endogenous Kv4 channels in rodent cardiomyocytes to test the hypothesis that PIAS3 is a SUMO E3 ligase for Kv4.2 and Kv4.3 channels and to examine cross talk between PIAS3 and PKA.
Materials and methods
Reagents
All chemicals and oligonucleotides were from Sigma unless otherwise indicated. All restriction enzymes were from NEB. All polymerases were from TAKARA. All secondary antibodies were from Jackson ImmunoResearch. Primary antibodies are indicated in Table 1. For siRNA experiments, siGenome Human UBE2I siRNA SMARTpool and non-targeting siRNA pool #2 were purchased from Dharmacon.
Cell Culture
Human Embryonic Kidney 293 (HEK293) cells were purchased from American Type Culture Collection (cat. no CRL-1573, lot number 2869494). HEK parental lines were cultured in Eagle’s Minimum Essential Medium (EMEM, Corning, cat. no 10009CV), supplemented with 10% Fetal.
Bovine Serum and 1% penicillin/streptomycin. The HEK-HCN2 cell lines, developed and validated as previously described using electrophysiology and western blot experiments [32], were cultured in the same media supplemented with 200 µM gentamicin.
Neonatal ventricular rat cardiomyocytes and culturing reagents were purchased from Lonza (cat. no R-CM-561). A vial of cardiomyocytes was resuspended by drop-wise addition of 9 mL of Rat Cardiomyocyte Growth Medium (RCGM). Cells were seeded at equal volumes onto 28, 15 mm laminin coated coverslips (Invitrogen cat. 23017015; 20 µg/mL). 4 h after seeding, 80% of the media was replaced with RCGM supplemented with 200 µM bromodeoxyuridine (BrdU) to limit fibroblast proliferation. For long-term culture, 50% of the media was removed and replaced with fresh RCGM + 200 µM BrdU every 3 days.
Plasmids, cloning, site-directed mutagenesis
A plasmid encoding a mouse Kv4.2 channel with GFP fused to the C-terminus (Kv4.2g) was kindly provided by Dr. Dax Hoffman [33]. Standard site-directed mutagenesis was used to mutate Kv4.2g-S552 into phosphodeficient (Kv4.2g-S552A) and phosphomimetic (Kv4.2g-S552E) plasmids using the PCR primers listed in Table 2. Sanger Sequencing of the entire open reading frame was used to verify that only intended mutations were generated (MCLAB). Kv4.2g-K579R, a SUMOylation-deficient mutant was previously created [26]. Because GFP contains putative SUMOylation sites, in some experiments GFP was removed from Kv4.2 g or its mutants using restriction digest with SalI and NotI to create a Kv4.2, GFP-null plasmid.
To generate a TAT-PIAS3 construct, a pCDNA3 TAT-HA backbone vector (Addgene, #14654) was digested with XhoI-SphI and dephosphorylated using standard techniques. The PIAS3 insert was constructed by amplifying a FLAG-PIAS3 template (Addgene, #152070) using PIAS3 forward and reverse primers (Table 2), followed by restriction digest with XhoI and Sph1. The PCR fragment was cloned into the XhoI-SphI restriction digested and phosphatased HA-TAT backbone vector using standard recombinant techniques. The complete open reading frame was verified with Sanger sequencing (MCLAB). This TAT-PIAS3 plasmid was then used to generate a TAT-PIAS3-ΔSIM1 plasmid. Standard site-directed mutagenesis experiments were performed to replace a portion of the SIM1 domain (VIDL) with four alanines using ΔSIM1 forward and reverse primers (Table 2). Sanger sequencing (MCLAB) of the entire open reading frame verified that only the intended mutation was created.
HA-KChIP2a and HA-DPP10c plasmids were constructed as previously described [27]. Additional plasmids purchased from Addgene included: SUMO2 (#17360), ubc9 (#14438), mCherry2-Cl (#54563), and Rab11aS25N (#46786). It is noted that the original FLAG-PIAS3 plasmid from Addgene contained a single nucleotide substitution (V580I) in the PIAS3 open reading frame that did not alter functionality [34].
Calcium phosphate transfections
HEK cells were seeded onto 60 mm dishes at ~ 70% density 24 h prior to transfection. Transfection media was prepared by resuspending DNA (10–25 µg) in TE Buffer (220 µl total volume); 30 µL of 2 M CaCl2 was then added; 250 µL of 2X HBS (275 mM NaCl, 10 mM KCl, 12 mM dextrose, 1.4 mM Na2HPO4, 40 mM HEPES, pH 7.05–7.10) was added dropwise with flicking. Once prepared, the transfection media was immediately added dropwise to the cells, which were then incubated at 37 °C in the CO2 incubator for 4 h to overnight before replacing with fresh media. Kv4.2 g, KChIP2a, and DPP10 were transfected at a molar ratio of 1:1:1 to form the ternary complex. Co-transfection experiments involving PIAS3 or mCherry (control) plasmids were performed at a 1:1 molar ratio (relative to ternary complex plasmids) for electrophysiology experiments and 5:1 molar ratio for immunoprecipitation (IP) and western blot (WB) experiments. siRNAs were used at 25 nM. Cells were either passaged to coverslips 24 h post-transfection and used the next day (electrophysiology) or lysed after 48–72 h (IP/WB experiments).
Generating non-denatured recombinant TAT-PIAS3 proteins
The TAT-PIAS3 or TAT-PIAS3-ΔSIM plasmid was transformed into BL21-CodonPlus (DE3)-RIPL Escherichia coli (Agilent Technologies), and 15% glycerol stocks were stored at -80 °C. Multiple induction protocols worked, but slow induction gave the best yield. A glycerol stock was streaked onto an agar plate. The next day, a fresh colony was inoculated into 250 ml of NZY broth with ampicillin and grown to O.D.600 = 1. The entire culture was diluted with 500 ml NZY broth + amp (total 750 ml) and grown to O.D.600 = 1. A 1 ml aliquot was saved at -20 °C and expression was induced by adding 10 ml, 100mM IPTG. The culture was grown overnight (16–18 h) at 22 °C with shaking. A 1 ml aliquot was stored at -20 °C, and bacteria were pelleted at 8,000xg for 20 min at 4 °C and resuspended in 8 ml native buffer (50mM NaH2PO4, 300mM NaCl, 20mM imidazole, pH 8); lysozyme was added to 1 mg/ml and the resuspension was incubated on ice for 30 min. The lysate was sonicated on ice. The sonicate was cleared by centrifugation at 10,000xg, for 35 min at 4 °C. The supernatant was transferred to a clean tube, mixed with 2 ml of equilibrated Nickel resin (Qiagen) and bound by rocking at 4 °C for 1 h. The slurry was added to a disposable Econo-Chromatography column (Bio-Rad). After settling, the column was washed once with 5 volumes of native buffer. The column was washed twice with 5 volumes of high salt native buffer (50mM NaH2PO4, 600mM NaCl, 20mM imidazole, pH 8). Flow-through and all wash fractions were saved. Recombinant peptides were eluted with 4 ml elution buffer (50mM NaH2PO4, 300mM NaCl, 250mM Imidazole), and 1 ml fractions were collected. An aliquot from each of the following fractions was run on a gel: uninduced culture, induced culture, flow through, all washes, each elution. The gel was stained with SYPRO Ruby; induction and elution of a peptide of the correct size was confirmed and non-specific contaminants were estimated (usually ~ 20% of the prep). Fractions containing the recombinant protein (usually fractions 1–3) were combined and desalted on a PD-10 column (GE Healthcare). The concentration of the isolated peptide was checked with a nanodrop (usually 100-200ng/µl) and confirmed with a western blot experiment using anti-HA antibody. The recombinant peptide was aliquoted and stored at -20 °C.
Electrophysiology
Whole cell patch clamp recordings were performed on HEK cells plated on poly-L-Lysine (100 µg/mL) coated coverslips. Cells were continuously superfused with extracellular saline (141 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose, 10 mM HEPES, pH 7.4, osmolarity ~ 300 mOsm/L). In some cases, cells were superfused with extracellular saline containing 8-bromo-cAMP (100 µM) during the recording for a maximum of 30 min. To measure the acute effects of PIAS3, transfected HEK cells were incubated with equal volumes of either PBS, TAT-PIAS3 (50 nM) or TAT-PIAS3-ΔSIM1 (50 nM) for 30 min at 37℃ and 5% CO2 prior to recording. Transfected cells were identified by GFP or mCherry expression and were patched with borosilicate glass pipettes (~ 2–3 MΩ) filled with intracellular saline (140 mM KCl, 1.2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, 2 mM MgATP, 10 mM HEPES, pH 7.2, osmolarity ~ 290 mOsm/L), and connected to a Multiclamp 700B Amplifier (Axon Industries). Whole cell capacitance was measured upon break-in. Fast and slow capacitance and series resistance were compensated. IA was recorded as previously described [35, 36]. The current was elicited with a 1s pre-pulse to − 90 mV followed by a depolarizing 250 ms test-pulse from − 50 mV to + 50 mV in 10 mV increments. The series was immediately repeated but the − 90mV prepulse was replaced with a pre-pulse to -30 mV to inactive Kv4.2 channels. Offline subtraction was performed to isolate IA. Steady state inactivation was measured using a protocol in which a 1.4 s voltage step from − 110 mV to -10 mV in 10 mV increments, was followed by test pulse to + 20 mV.
Ih was elicited from HEK cells stably expressing an HCN2-GFP fusion protein (HEK-HCN2) using a series of 5s hyperpolarizing voltage steps from − 50 mV to -120 mV in 10 mV increments. Steady state peak current was measured by subtracting the initial fast leak current from the slowly activating Ih current at the end of each voltage step.
For IA and Ih, the maximal conductance (Gmax) and the voltage of half-activation (V1/2 act) were determined by plotting conductance against the voltage and fitting the data with a first-order Boltzmann equation. For IA, the fast (τf) and slow (τs) time constants of inactivation were determined by fitting the decay current for the + 50 mV test pulse with a two-term exponential equation. For IA, the V1/2 of inactivation was determined by plotting the current against the voltage and fitting to a first-order Boltzmann equation.
Immunoprecipitation
72 h post transfection, HEK cells were lysed in RIPA buffer (1% NP40, 50mM Tris-HCl pH7.4, 150mM NaCl, 0.1% SDS, 0.5% DOC, 2mM EDTA, 20mM NEM, 1:100 protease inhibitor) for 30 min at 4℃. Lysates were collected and debris was pelleted by centrifugation for 10 min at 14,000 rpm. The protein concentrations of the resulting supernatants were determined using the Pierce BCA Assay kit. Equal concentrations of lysates were incubated with a Guinea Pig anti-Kv4.2 or Guinea Pig IgG antibody overnight at 4℃ (5 µg antibody/mg lysate). Immunoprecipitations were performed using the Pierce Classic Magnetic IP/Co-IP Kit according to manufacturer’s instructions. IP elutions were conducted in Pierce low pH Elution Buffer at 100 µL/mg of protein. The eluant was immediately neutralized with Pierce neutralization buffer at a ratio of 1:10. For any given set of +/- PIAS3 transfections, the resulting IPs were always transferred onto the same membrane to ensure intra-assay conditions were the same regardless of treatment group. This is demonstrated in Supplemental Figs. 1 and 2. Run conditions, transfer times, and incubation times were held constant to minimize inter-assay variability. A similar protocol was used in Kv4.2 co-IP experiments with mouse heart lysates. In these experiments, wild-type, male and female mouse hearts were dissected, and flash frozen at -70 °C. One heart was pulverized in liquid nitrogen using a mortar and pestle, and then resuspended in RIPA buffer, and used to generate protein lysates as described above. Immunoprecipitations using the Pierce Classic Magnetic IP/Co-IP Kit was as described above.
Western blot (WB) assays
To determine the level of Kv4.2 SUMOylation, Kv4.2 IP products were fractionated on duplicate gels using SDS-PAGE (20µL of IP eluant/ lane) and then transferred onto two PVDF membranes using a wet electroblotting system (BioRad). The membranes were blocked in 5% non-fat milk in TBS (50 mM Tris-HCl pH7.4, 150 mM NaCl) for 1-3 h at room temperature, and then incubated with a primary antibody against rabbit anti-SUMO1 or rabbit anti-SUMO2 in 1% T-TBS solution (TBS + 0.1% Tween20) overnight at 4℃. The next day, membranes were washed and incubated with an Alkaline Phosphatase conjugated secondary antibody in 1% T-TBS solution for 2 h at room temperature. Membranes were washed and incubated with AP substrate (Bio-Rad) and the chemiluminescent signal imaged with Axion Biosystems 6000. Membranes were then stripped using a mild stripping buffer (200 mM glycine, 0.1% SDS, and 1% Tween 20, pH 2.2) and re-probed with a mouse anti-Kv4.2 primary antibody followed by imaging. ImageJ was used for WB analysis. The optical density (OD) of the SUMO signal was divided by that of the OD for Kv4.2. For KChIP2a and DPP10 co-IP experiments, Kv4.2 IP products were used and treated as stated above. IP products were probed first with either mouse anti-SUMO1 or rabbit anti-SUMO2 and then stripped and re-probed with mouse anti-pan-KChIP or mouse anti-HA.
A similar protocol was used to identify Kv4.2-PIAS3 interactions in mouse heart lysates except signals were not quantified (n = 3 hearts for SUMO and 3 hearts for PIAS3 experiments).
To determine the knockdown of Ubc9 in siRNA lysates, cells were lysed in RIPA buffer and lysates were prepared as described for the IP experiments above. The siRNA lysates were used to generate western blots as described above. Both experimental (siUbc9 treatment) and control (siScrambled treatment) lysates were run on the same gel. The blot was stained with SYPRO RUBY protein blot stain (Bio-Rad) and imaged to determine the total protein OD for each lane. The blot was then sequentially probed with rabbit anti-ubc9 and an alkaline-phosphatase-conjugated anti-rabbit secondary as described above. After imaging and measuring the OD, the Ubc9 signal was divided by the total protein signal.
Immunofluorescence
TAT-PIAS3 or TAT-PIAS3-ΔSIM1 in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) or vehicle (PBS alone) were bath-applied to HEK cells in 24 well plates and incubated for 30 min at 37 °C and 5% CO2. TAT-tagged proteins were used at a final concentration of ~ 50 nM. Cells were washed with 1x PBS and fixed in the dish with 4% paraformaldehyde in PBS for 15 min at room temperature. Cells were permeabilized using 3, 5 min washes with 1x PBS + 0.05% Triton X-100 at room temperature. Cells were blocked for 1 h in 1% Bovine Serum Albumin (BSA), 0.3 M glycine in PBST (PBS + 0.1% Tween20) at room temperature. Cells were then incubated with mouse anti-TAT primary antibody + 1% BSA + 5% normal donkey serum (NDS) in PBST for 1 h at room temperature. Cells were washed 3x, 5 min each with 1x PBST at room temperature. Cells were incubated with donkey anti-mouse AlexaFluor 568 secondary antibody in PBST + 5% NDS for 1 h at room temperature. Cells were then washed 3x, 10 min each with 1x PBS. The second wash contained 300nM DAPI. Cells were imaged in the dish using a Keyence BZ-X series fluorescent microscope and analyzed with ImageJ. To obtain pixel intensities for the red channel, the rolling ball method was first used to subtract background from a given red micrograph. Regions of interest (ROI) were then designated by outlining large clusters of cells (5–7 ROIs/micrograph). The mean pixel intensity was determined for each ROI, and values were averaged to obtain the mean pixel intensity for the entire micrograph. To obtain representative images, red and blue micrographs were merged, and background was subtracted with the rolling ball method.
Proximity ligation assay
Cardiomyocytes (Lonza) were subjected to 1 of 5 bath-applied treatments for 30 min at 37℃ and 5% CO2: PBS, TAT-PIAS3 (50 nM), TAT-PIAS3-ΔSIM1 (50 nM), 8-bromo-cAMP (100 µM) or 8-bromo-cAMP (100 µM) + TAT-PIAS3 (50 nM). Next, coverslips were washed with 1x PBS for 2 min and then fixed with a 4% PFA solution for 10 min. Coverslips were then washed with 1x PBS for 2 min before incubation with WGA-CF®594 (Biotium Aldrich 3.3 ug/mL) for 10 min. Coverslips were washed with 1x PBS for 2 min and then permeabilized with 1x PBS with 0.05% Triton X-100 3x at 5-minute intervals. Coverslips were blocked in 35 mm dishes with Duolink blocking solution for 30 min at 37℃. After the block, coverslips were incubated with primary solutions overnight at 4℃. Primary solutions contained either rabbit Kv4.2 (1:400) or rabbit Kv4.3 (1:400) and mouse SUMO1 (1:100) and mouse SUMO2 (1:70) antibodies diluted in Duolink antibody diluent. The next day, coverslips were washed with 1x PBS 3x at 5-minute intervals, and then 2x in Duolink Wash Buffer A. Coverslips were incubated with Duolink rabbit plus and mouse minus probes diluted 1:5 in Duolink antibody diluent for 1 h at 37℃. Coverslips were washed 2x in Wash Buffer A and then incubated with the Duolink ligase reaction for 30 min at 37℃. Coverslips were washed 2x in Wash Buffer A, and then incubated with the Duolink amplification reaction for 2 h at 37℃. Coverslips were washed 2x in Wash Buffer B for 10 min, 1x in 0.01x Wash Buffer B for 1 min, and then incubated with DAPI (300 nM) for 5 min. Coverslips were washed with 1x PBS and mounted with VectaShield. The experiment was repeated ≥ 2X times for each treatment group using a different vial of cells (i.e., different heart) for each replicate. Monolayers were imaged with a Keyence BZ-X series fluorescent microscope; ≥30 non-overlapping images were obtained per coverslip. TIFF files were analyzed with ImageJ. A cell was defined by the presence of at least one DAPI-stained nucleus and a clear perimeter. A line was drawn around the outline of all cells in a micrograph. This was used to record the surface area for each cell. Using the red channel, the size and intensity of each punctum (raw integrated density, RID) in a cell was measured and masks were generated using the particle analysis program. Masks were merged onto micrographs and inspected for accuracy. If > ~ 10% of the signal was not represented in the mask (approximated by eye), then the cell was excluded from further analysis. Fewer than 10 cells were excluded across all treatment groups. The RIDs for each cell were summed and recorded (termed SUMOylation index) and divided by the surface area of the cell to yield a value termed the normalized SUMOylation index for that cell. Normalized SUMOylation indices for all cells within a treatment group were compared across treatment groups. Experimenters were blinded for the imaging and analysis portions of the experiment.
Statistical analyses
GraphPad PRISM 9 software was used for statistical analysis. Normality and homogeneity of variance were assessed for each data set. Data points > 2 standard deviations from the mean were considered outliers and were excluded, except for the PLA studies where all data points were included, and outliers were indicated as symbols in Tukey box plots. In all cases, the significance threshold was set at p < 0.05. Parametric data were analyzed with either unpaired t-tests, one-way ANOVAs, or Welch’s ANOVA (for unequal variances) followed by the indicated post hoc tests when appropriate. Non-parametric data were analyzed with either a Mann-Whitney U test or a Kruskal-Wallis test with Dunn’s multiple comparisons post hoc. The results of each statistical test performed (if any) are listed in every Figure legend.
Results
PIAS3 overexpression mimics and occludes the effect of SUMO2 + ubc9 overexpression on IAGmax in HEK293 cells
The Kv4.2g ternary complex (TC) studied herein comprises a Kv4.2-GFP fusion protein (Kv4.2g), HA-KChIP2a and HA-DPP10c. We previously demonstrated that co-expressing SUMO2 and ubc9 with the Kv4.2g TC in HEK293 cells enhanced Kv4.2g-K579 SUMOylation, which in turn, produced a significant ~ 30–50% rise in Kv4.2 surface expression, a significant 37–70% increase in IA maximal conductance (Gmax), and a 65% decrease in channel internalization [27]. Recent work showed that rab11a, which regulates slow recycling, was necessary for the effects of SUMOylation on Kv4.2 channels [28]. If PIAS3 is a SUMO E3 ligase for Kv4 channels, then co-expressing PIAS3 should mimic and occlude the effect of co-expressing SUMO2 + ubc9. To test this hypothesis, whole cell patch clamp recording was performed on HEK cells transiently transfected with plasmids encoding the Kv4.2g TC with or without a plasmid for PIAS3, or plasmids for PIAS3 + SUMO2 + Ubc9 (Fig. 1).

PIAS3 mimics and occludes the effect of Kv4.2 SUMOylation. IA was characterized using whole cell patch clamp recordings on HEK cells expressing the Kv4.2g TC. A Representative experiments for each treatment group show current (top panel) and voltage (bottom panel) traces from the activation protocol. B PIAS3 overexpression mimicked and occluded the effect of SUMO + ubc9 overexpression. Bar graphs show mean IA Gmax ± SEM. Each symbol represents one cell. Data were obtained from ≥ 3 independent transfection experiments. The mean IA Gmax for the PIAS3 treatment group (139.0 ± 15 nS) was significantly greater than the control (87.7 ± 6.2 nS), while co-expression of SUMO and ubc9 produced no further effect (176.3 ± 16.3 nS). *, significantly different using a one-way ANOVA with a Tukey’s post hoc test that makes all pairwise comparisons, F(2,32) = 18, p = 0.0001; NS, not significant. C Representative experiments show current (top panel) and voltage traces (bottom panel) from the steady state inactivation protocol. D Mean normalized activation (circle) and steady state-inactivation (square) curves for each treatment group. Each symbol represents the mean ± SEM for all cells in a treatment group, n for each treatment group is the same as shown in B. Data were obtained from ≥ 3 independent transfection experiments. There was no significant difference in the act V1/2 between treatment groups as determined with a one way ANOVA F(2,33) = 2.094, p = 0.139. PIAS3 induced a significant 8mV shift in the inact V1/2 relative to the other treatment groups as determined using a one way ANOVA followed by a Tukey’s post hoc test that makes all pairwise comparisons, F(2,33) = 5.712, p = 0.0074
PIAS3 overexpression mimicked the effect of SUMO + ubc9 on IA Gmax (Fig. 1A and B). It significantly increased IA Gmax by ~ 60% compared to the Kv4.2g TC alone (Fig. 1B). Additionally, PIAS3 overexpression occluded the effect of SUMO + ubc9; PIAS3 + SUMO + ubc9 did not significantly increase IA Gmax relative to PIAS3 alone (Fig. 1B). Overexpression of SUMO2 + Ubc9 did not significantly alter the fast (τf) or slow (τs) time constants of inactivation [27], and neither did overexpression of PIAS3 (τf, 22 ± 2ms vs. 29 ± 3ms; τs, 73 ± 5ms vs. 81 ± 11ms, t-test, p > 0.05). Overexpression of SUMO2 + ubc9 did not significantly alter the voltage of half activation (act V1/2) [27], and neither did overexpression of PIAS3 (-5.3 ± 6.2mV vs. -6.6 ± 2.2mV) (Fig. 1A and D). Overexpression of SUMO2 + ubc9 did not alter steady-state inactivation [27]. In contrast, PIAS3 overexpression produced a significant ~ 8mV depolarizing shift in the voltage of half inactivation (inact V1/2) that increased the window current (Fig. 1C&D). This shift was prevented when PIAS3 was overexpressed with SUMO2 + Ubc9. Since PIAS3 affects a subset of the SUMO sites targeted by ubc9, the simplest interpretation is that a site targeted by ubc9, but not by PIAS3, produced an effect that obscured/prevented the shift in inact V1/2.
We previously demonstrated that Kv4.2 possessed at least two SUMOylation sites: K437 and K579 [26]. The SUMOylation induced increase in IA Gmax, mediated by the Kv4.2g TC in HEK293 cells, was due to enhanced SUMOylation at Kv4.2-K579 [27]. Overexpression of SUMO2 + ubc9 had no effect when the ternary complex contained the SUMOylation-deficient α-subunit, Kv4.2-K579R; however, enhanced SUMOylation still increased channel surface expression and IA Gmax when the ternary complex contained the SUMOylation-deficient α-subunit, Kv4.2-K437R [27]. To determine if PIAS3 also acted at Kv4.2-K579, the whole cell patch clamp experiments were repeated on HEK cells expressing a SUMOylation-deficient Kv4.2g-K579R TC (Fig. 2). The mutation blocked the PIAS3-induced increase in IA Gmax (Fig. 2B) and the shift in the inact V1/2 (Fig. 2C). These data indicated that the previously identified SUMOylation site, Kv4.2-K579, was necessary for PIAS3-mediated alterations in IA.

PIAS3 requires K579 for its effect on Kv4.2 channels. IA was characterized using whole cell patch clamp recordings on HEK cells expressing a SUMOylation deficient Kv4.2g-K579R TC. A Representative experiments show current traces from the activation (left) and steady-state inactivation (right) protocols. B PIAS3 has no effect on IA Gmax relative to control for the SUMO-deficient channel (102.2 ± 7.4nS vs. 116.8 ± 11.8nS, t-test, p = 0.287); NS, not significant. Bar graphs show mean IA Gmax ± SEM. Each symbol represents one cell. Data were obtained from ≥ 3 independent transfection experiments. C Mean activation (circle) and steady state-inactivation (square) curves for each treatment group. Each symbol represents the mean ± SEM for all cells in a treatment group, n for each treatment group is the same as in B. Data were obtained from ≥ 3 independent transfection experiments. There was no significant difference between treatment groups for act V1/2 (-2.7 ± 1 mV vs. -3.1 ± 1.7 mV; t-test, p = 0.827) or inact V1/2 (-54.4 ± 0.6 mV vs. -56.3 ± 1.6 mV; t-test, p = 0.184)
Rab11a is a small GTPase that critically regulates several aspects of transmembrane protein recycling [37]. Studies on the Kv4.2g TC in HEK cells revealed that rab11a was necessary for the SUMOylation-induced increase in IA Gmax [28]. Rab11a-S25N is a dominant-negative mutant that is incapable of binding GTP, which inactivates rab11a and prevents delivery of proteins to the recycling endosome [38]. If the PIAS3-induced increase in IA Gmax relies on enhanced recycling of endocytosed channels, then the dominant negative mutant should block the effect of PIAS3. HEK cells were transfected with the Kv4.2g TC with or without PIAS3 and with or without Rab11aS25N. Co-expression of Rab11aS25N with the Kv4.2g TC had no significant effect on IA Gmax compared to the Kv4.2g TC alone, but it blocked the PIAS3-mediated increase in IA Gmax (Fig. 3). These results suggest that the PIAS3-mediated increase in IA Gmax is due to enhanced recycling of endocytosed channels.

Rab11a is necessary for the PIAS3 induced increase in IAGmax. Whole cell patch clamp experiments were performed on HEK cells transiently expressing the Kv4.2g TC +/- PIAS3 and +/- rab11aS25N. The mean ± SEM IA Gmax was plotted for each treatment group. Each symbol represents one cell. Data were obtained from ≥ 3 independent transfection experiments. The data for cells lacking rab11aS25N were replotted from Fig. 1 for comparison. Rab11aS25N alone had no effect on IA Gmax relative to control, but it blocked the PIAS3-mediated increase in IA Gmax. *, significantly different, one-way ANOVA with Tukey’s post hoc test that makes all pairwise comparisons, F(3,40) = 7.1, p = 0.0006; NS, not significant
In sum, the data show that when the Kv4.2g TC is expressed in HEK cells, overexpression of PIAS3 mimics and occludes the effects of enhanced SUMOylation at Kv4.2-K579. This is consistent with the hypothesis that PIAS3 is a SUMO E3 ligase for Kv4 channels.
Catalytic activity and ubc9 are necessary for the PIAS3-mediated increase in IAGmax
If PIAS3 acts as a SUMO E3 ligase for Kv4 channels, then only a catalytically active PIAS3 should produce an increase in IA Gmax. The SIM1 domain is necessary for PIAS3 catalytic activity [17]. The next experiment tested if bath-application of a membrane-permeable PIAS3 could enhance IA Gmax when SIM1 was mutated (Fig. 4).

An active, but not a catalytically inactive, PIAS3 mimics the acute effect of an increase in SUMO on IAGmax. A Recombinant, membrane permeable PIAS3 proteins were generated: TAT-PIAS3 (wild type) and TAT-PIAS3-ΔSIM1 (catalytically inactive). The diagram shows TAT-PIAS3 constructs. Flag-PIAS3 was subcloned into a TAT-HA-HIS vector. The conserved domains of PIAS3 include SAP, PINIT, SP-RING, SIM1, and SIM2. SIM1 orients the SUMO peptide for attachment to the target, and a wild-type SIM1 domain is necessary for PIAS3-mediated SUMOylation. The wildtype SIM1 (blue) domain of TAT-PIAS3 was mutated (green) to produce a catalytically inactive PIAS3 (ΔSIM1). Recombinant proteins were isolated using Nickle columns with BL21 cell lysates. B Immunofluorescence experiments verified that bath-applied, TAT-tagged proteins crossed the plasma membrane and showed similar cytosolic distributions. TAT-PIAS3 (~ 50nM), TAT-PIAS3-ΔSIM1 (~ 50nM) or vehicle (PBS) were bath-applied (30 min, 37 °C); cells were fixed, stained with DAPI (blue) and for the TAT-tag (red), and imaged with a Keyence microscope. A representative merged image from each treatment group, obtained with the same microscope settings, is shown. Note the color of the nuclei varies with the amount of red staining in the merged image. All scale bars are 20 μm. C Cell penetration was quantified for the 3 treatment groups shown in B using ImageJ. Mean pixel intensities were measured for the red channel as described in Materials and Methods. Each symbol represents the average red pixel intensity for 1 micrograph. *, significantly different as determined using a Welch’s ANOVA with Dunnett’s 3T post hoc test that makes all pairwise comparisons, W(2, 22.74) = 572.9, p < 0.0001. D SUMO peptides were or were not added to the recording pipet during whole cell patch clamp experiments. Acute application of SUMO peptide (4.2µM) significantly increased IA Gmax. Data were taken from [27] for the sake of comparison. E HEK Kv4.2g TC cells were incubated with PBS (control), TAT-PIAS3 (50nM), or TAT-PIAS3-ΔSIM1 (50nM) for 30 min at 37℃ and 5% CO2 prior to whole cell recording. Acute application of TAT-PIAS3, but not TAT-PIAS3-ΔSIM1, produced a significant increase in IA Gmax. Bar graphs show the mean ± SEM IA Gmax. Each symbol is one cell. All data were normalized by the mean for the PBS treatment group. Data were from ≥ 3 replicate experiments. *, significantly different as determined using a one way ANOVA with Tukey’s post hoc test that makes all pairwise comparisons, F(2, 15) = 20.7, p < 0.0001; NS, not significant
A recombinant, membrane-permeable PIAS3 protein (TAT-PIAS3) was generated by cloning Flag-tagged PIAS3 into an N-terminal TAT-HA-HIS vector (Fig. 4A). This construct was used to generate a catalytically deficient PIAS3 (TAT-PIAS3-ΔSIM1) where key amino acids in the SIM1 domain were replaced with alanine as previously described [18]. To confirm that the TAT-tag conferred cell-penetrating properties on the ~ 65kD PIAS3 proteins [39], a TAT-tagged protein (50nM) or vehicle (PBS) was bath applied to HEK cells. Cells were fixed and used in immunofluorescence experiments to visualize the TAT-tag (red) and cell nuclei (blue). Representative merged micrographs (Fig. 4B) showed that red immunofluorescence was absent in the PBS treatment group but was similarly distributed in the TAT-PIAS3 and TAT-PIAS3-ΔSIM1 treatment groups. The level of cell penetration was also highly similar for the two recombinant proteins (Fig. 4C).
Welch et al. [27] previously showed that IA Gmax could be similarly enhanced by acute and long-term increases in SUMO. Including SUMO peptides (4.2µM) in the recording pipet increased IA Gmax by ~ 60% (Fig. 4C). Similarly, a 30 min bath application of TAT-PIAS3 (50nM) but not TAT-PIAS3-ΔSIM1 (50nM) produced a significant ~ 60% increase in IA Gmax in HEK cells expressing the Kv4.2g TC (Fig. 4D). These data indicated that the acute effects of SUMO and PIAS3 on IA Gmax were similar, and that the SIM1 domain of PIAS3 was necessary for the PIAS3-induced increase in IA Gmax.
To determine if the sole SUMO-conjugating enzyme, ubc9, was necessary for the PIAS3-induced increase in IA Gmax, cells were patch clamped after siRNA knock down of ubc9. HEK cells were transfected with plasmids encoding the Kv4.2g TC with or without PIAS3 and with siRNA for either ubc9 (siUbc9, 25 nM) or a control (siScramble, 25 nM). For each transfection, cells were passaged to a 35 mm dish for western blot analysis (Fig. 5A-B) or coverslips for whole cell patch clamp recording (Fig. 5C). Relative to siScramble, siUbc9 decreased ubc9 expression by an average 65 ± 8%. Whole cell patch clamp recordings showed that the PIAS3-induced increase in IA Gmax was observed only for the siScramble but not the siUbc9 treatment group (Fig. 5C). Thus, ubc9 was necessary for the PIAS3-induced increase in IA Gmax.

The SUMO-conjugating E2 enzyme, ubc9, is necessary for the PIAS3-induced increase in IAGmax. HEK cells were transfected with siRNA targeting ubc9 (siUbc9) or a scrambled control (siScramble) along with plasmids encoding Kv4.2g TC +/- PIAS3. For each transfection, cells were plated onto both a 35 mm plate for western blot experiments (A-B) and onto coverslips for whole cell patch clamp recordings (C). A Representative experiment showing the effect of siUbc9. SDS-PAGE gel containing cell lysates were transferred to PVDF membranes, which were then stained with Sypro-Ruby (right) to measure total protein concentration followed by a western blot experiment to measure ubc9 (left). For each lysate, ubc9 expression was quantified by dividing the optical density (O.D.) for ubc9 by the O.D. for total protein. B Treatment with siUbc9 produced a ~ 65% knock down relative to treatment with siScramble. Ubc9 expression for each lysate was divided by the mean for the scrambled control. Normalized Ubc9 expression was plotted. Each symbol in the plot represents one independent lysate. Bar graphs show the mean ± SEM. *, significant difference determined with a t-test, p < 0.0001. C Plots of normalized IA Gmax from whole cell patch-clamp experiments showed that ubc9 knock down blocked the PIAS3-induced increase in IA Gmax while the scrambled control had no effect on the ability of PIAS3 to elicit an increase in IA Gmax. *, significant; ns, non-significant; one way ANOVA with Tukey’s post hoc test that made all pairwise comparisons F(3,20) = 4.976, p = 0.0001
Together these experiments suggest that PIAS3 increases IA Gmax by catalyzing the addition of SUMO to Kv4 channels.
PIAS3 increases Kv4.2-K579 SUMOylation
Lastly, to test if PIAS3 acts as a bona fide SUMO E3 ligase for Kv4.2-K579, we used immunoprecipitation (IP) and western blot experiments to measure SUMOylation in the Kv4.2 and Kv4.2-K579R TC with and without overexpression of PIAS3 (Fig. 6 and Supplemental Fig. 1). For these experiments, GFP tags were removed from both Kv4.2 constructs since GFP, itself, contained putative SUMOylation sites.

Increasing PIAS3 expression results in enhanced SUMOylation at Kv4.2-K579. A Representative results for IP followed by western blot experiments using protein lysates from HEK cells expressing Kv4.2 TC with or without PIAS3. IP and western blot (WB) antibodies were as indicated. Western blots were first probed with anti-SUMO, imaged, stripped, and re-probed with anti-Kv4.2. B Bar graphs show the mean normalized fraction of SUMOylated Kv4.2 ± SEM. The O.D. for each signal was measured. The SUMO O.D. was divided by the Kv4.2 O.D. to obtain the fraction of SUMOylated channels for a given lysate. All data points were then divided by the mean for the control treatment group. Each symbol in the graph represents an independent transfection and IP/western blot experiment. *, significantly different, ns, not significant (SUMO1: 1.00 ± 0.2 vs. 2.7 ± 0.4; t-test, p = 0.021; SUMO2: 1.00 ± 0.29 vs. 1.2 ± 0.18; t-test, p = 0.5032). C Representative results for IP followed by western blot experiments using protein lysates from HEK cells expressing Kv4.2-K579R TC with or without PIAS3. IP and western blot antibodies were as indicated. The protocol was as described in A. D Bar graphs show the mean normalized fraction of SUMOylated Kv4.2-K579R ± SEM. Measurements and analyses were as in B. ns, not significant (SUMO1: 1.00 ± 0.3 vs. 1.05 ± 0.34, t-test, p = 0.9146; SUMO2: 1.00 ± 0.32 vs. 0.89 ± 0.38, t-test, p = 0.8373)
PIAS3-induced a significant 2.7-fold increase in the level of endogenous SUMO1ylation of Kv4.2 but no change in the level of SUMO2ylation (Fig. 6B). This is not unusual as SIM domains can exert selective preference for a specific SUMO isoform [40, 41].
Experiments were repeated with the SUMOylation deficient Kv4.2-K579R TC. The mutant channel was SUMOylated under control conditions (Fig. 6C), suggesting that there is more than one SUMOylation site on Kv4.2 channels, as we previously demonstrated [26]. The non-Kv4.2-K579 site(s) appeared to account for most Kv4.2 SUMOylation under baseline conditions, as the level of Kv4 SUMOylation was highly similar when comparing wild type and mutant control groups. PIAS3 overexpression produced no significant change in the SUMOylation of Kv4.2-K579R channels (Fig. 6D). These data are consistent with our hypothesis that PIAS3 increases SUMOylation at Kv4.2-K579.
In sum, the experiments performed thus far on Kv4.2g TC in HEK cells show that (1) PIAS3 mimics and occludes the effect of enhanced SUMOylation on IA Gmax, (2) the previously identified SUMOylation site, Kv4.2-K579, is necessary for PIAS3 effects on IA Gmax, (3) the SUMOylation machinery (E2 enzyme and a catalytically active E3) is necessary for the PIAS3-induced increase in IA Gmax and (4) PIAS3 increases SUMOylation at Kv4.2-K579. These data are compelling; together they demonstrate that PIAS3 is a bona fide SUMO E3 ligase for Kv4 channels. It acts at Kv4.2-K579 to increase rab11-dependent recycling of endocytosed channels, thereby increasing surface expression and IA Gmax.
PKA-mediated phosphorylation at Kv4.2-S552 blocks PIAS3-mediated SUMOylation at Kv4.2-K579 and the attendant increase in IAGmax
Phosphorylation plays a permissive role in SUMOylation, and Kv4.2-K579 is a SUMOylation island in a sea of phospho-regulation (Fig. 7A). Mass spectrometry on protein isolates from cultured neurons previously identified eleven Kv4.2 phosphorylation sites [42], ten of which clustered around K579. The short segment of 68 amino acids comprising these sites (552–620) is identical in mouse, rat and human Kv4.2 proteins. One possibility is that this conserved fragment represents a module for integrating parallel signals to influence a binary output (K vs. K + SUMO) that controls the recycling rate of a population of channels. One site, Kv4.2-S552, was previously shown to decrease Kv4.2 surface expression in neurons [43, 44]. If our hypothesis is correct, then phosphorylation at Kv4.2-S552 should block SUMOylation at Kv4.2-K579 and thereby reduce recycling and surface expression of Kv4.2 channels. To begin to evaluate this idea, HEK cells were transiently transfected with HA-DPP10c + HA-KChIP2a + Kv4.2g with or without PIAS3. After 48 h, IA was analyzed with whole cell patch clamp with or without bath application of 8-bromo-cAMP, a membrane permeable cAMP analog that activates PKA. Bath-applied 8-bromo-cAMP had no effect on IA Gmax by itself, which is consistent with the idea that Kv4.2-K579 is not SUMOylated under baseline conditions (see above discussion of Fig. 6). The increase in intracellular cAMP blocked the PIAS3-mediated increase in IA Gmax (Fig. 7B). To assess if inhibition of the PIAS3 effect was due to PKA-mediated phosphorylation at Kv4.2-S552, site-direct mutagenesis was used to create phosphodeficient (Kv4.2g-S552A) and phosphomimetic (Kv4.2g-S552E) constructs. Whole-cell patch clamp experiments with the phosphodeficient Kv4.2g-S552A TC showed that 8-bromo-cAMP could no longer prevent a PIAS3-mediated increase in IA Gmax (Fig. 7C). Thus, PKA-mediated phosphorylation at Kv4.2-S552 was necessary to block the PIAS3 effect. Experiments using the Kv4.2g-S552E TC showed that the phosphomimetic mutation mimicked and occluded the effect of bath-applied 8-bromo-cAMP. PIAS3 did not produce an increase in IA Gmax and superfusion with 8-bromo cAMP had no further effect (Fig. 7D). Thus, PKA-mediated phosphorylation at Kv4.2-S552 was sufficient to block the PIAS3-mediated increase in IA Gmax. Sufficiency was confirmed by measuring SUMOylation at Kv4.2-K579 in the phosphomimetic TC (Fig. 8). HEK cells were transiently transfected with plasmids encoding the Kv4.2-S552E TC with and without PIAS3. Note GFP was removed from Kv4.2-S552E. After 48 h, cells were lysed and used in IPs with anti-Kv4.2 antibody followed by western blot experiments. The fraction of SUMOylated channels was determined. PIAS3 did not enhance SUMO1 (Fig. 8A&B) or SUMO2 (Fig. 8C&D) decorations at Kv4.2-K579. Together, the data presented thus far provide compelling evidence that PKA-mediated phosphorylation at Kv4.2-S552 blocks PIAS3-mediated SUMOylation at Kv4.2-K579 and the accompanying increase in IA Gmax.

PKA-mediated phosphorylation at Kv4.2-S552 blocks PIAS3-mediated SUMOylation at Kv4.2-K579. A Diagram of the conserved 68 amino acid Kv4.2 fragment comprising the K579 SUMOylation site surrounded by 10 proximal phosphorylation sites identified with mass spectrometry. Phosphorylation sites that were previously investigated for their effects on Kv4.2 are indicated with a star. B-D HEK cells expressing the indicated TC +/- PIAS3 were or were not superfused with 100 µM 8-bromo-cAMP, a cell permeable activator of PKA, and whole cell patch clamp was performed to measure IA Gmax. Data were obtained from ≥ 3 transfections. All data points were divided by the mean for the control group (no PIAS3, no cAMP). Bar graphs show the normalized mean IA Gmax ± SEM. Each symbol represents one cell; *, significantly different; ns, non-significant. In B, the significant PIAS3-induced increase in IA Gmax was blocked by 8-bromo-cAMP in HEK cells expressing the Kv4.2g TC (one-way ANOVA with Tukey’s post hoc test that makes all pairwise comparisons, F(3,53) = 8.877, p < 0.0001). In C, PIAS3 was able to produce a significant increase in IA Gmax in the presence of 8-bromo-cAMP in HEK cells expressing the Kv4.2g-S552A TC (one-way ANOVA with Tukey’s post hoc test that makes all pairwise comparisons F(2,36) = 8.343, p = 0.001). In D, PIAS3 did not induce an increase in IA Gmax, and 8-bromo-cAMP had no effect on HEK cells expressing the Kv4.2-S552E TC (one-way ANOVA F(3,43) = 1.03, p = 0.3887)

PKA-mediated phosphorylation at Kv4.2-S552 blocks the PIAS3-mediated increase in Kv4 SUMOylation. A WB of Kv4.2 SUMO1 conjugation. Blots containing Kv4.2 and IgG IPs using protein lysates from HEK cells expressing Kv4.2-S552E TC +/- PIAS3 were probed with anti-SUMO1 and then stripped and re-probed with anti-Kv4.2 (~ 65kD). B The fraction of SUMOylated channel (SUMO OD÷Kv4 OD) was normalized to control (no PIAS3) and plotted. Bar graphs show the mean normalized fraction of SUMOylated Kv4.2 ± SEM. Each data point represents a separate transfection/IP-western blot experiment. The phosphomimetic mutation blocked the PIAS3-mediated increase in Kv4 SUMO1 conjugation seen in Fig. 6 (1.00 ± 0.13 vs. 0.85 ± 0.17, Mann-Whitney U, p = 0.377). C-D Experiments were repeated to measure SUMO2 conjugation. PIAS3 did not alter SUMO2 conjugation compared to the Kv4.2-S552E TC alone (1.00 ± 0.28 vs. 0.70 ± 0.14, t-test, p = 0.37)
PIAS3-mediated SUMOylation of cardiomyocyte Kv4 channels can be blocked by an increase in cAMP
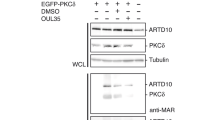
The interaction between Kv4.3 and PIAS3 was first discovered using co-IP experiments on rat heart lysates [22]. The aforementioned Kv4 SUMOylation and phosphorylation sites are conserved across Kv4.2 and Kv4.3 isoforms and across species. We used co-IP followed by western blot experiments with mouse heart lysates to confirm that Kv4.2 channels were SUMOylated (Fig. 9A) and to detect an interaction between Kv4.2 and PIAS3 (Fig. 9B). We then asked if increasing PIAS3 could enhance Kv4 channel SUMOylation in rat cardiomyocytes. Neonatal rat ventricular cardiomyocytes (Lonza) were cultured on coverslips. Cells received a 30 min bath-application of TAT-PIAS3 (50nM) or TAT-PIAS3-ΔSIM1 (50nM) or vehicle (PBS). Proximity ligation assays (PLA) were then used to measure Kv4 channel SUMOylation. PLAs were performed with anti-SUMO and anti-Kv4.3 or anti-Kv4.2 antibodies using a Duolink in situ PLA kit (Sigma). Cells were imaged with a Keyence microscope (Fig. 9C top panel). A punctate signal was created when SUMO and Kv4 were < 40 nm apart, i.e., the Kv4 channel was SUMOylated. SUMOylated channels were predominately located in a perinuclear region, although they could also be observed to a lesser extent throughout the cell and along the periphery. SUMOylation was quantified with ImageJ. All cells within a field of view were outlined. The size and intensity of each punctum (raw integrated density, RID) in all outlined cells were quantified using Image J particle analysis. Masks showing quantified RID were created for each cell outlined in yellow (Fig. 9C bottom panel). Masks were merged onto micrographs and inspected to ensure they were representative. The RIDs for all puncta in a cell were summed; this was termed the SUMOylation index for the cell. The SUMOylation index was divided by the surface area of the cell (encompassed by yellow line) to yield the normalized SUMOylation index for a cell. Plots of Kv4.3 (Fig. 9D) and Kv4.2 (Fig. 9E) normalized SUMOylation indices for each treatment group showed that TAT-PIAS3 produced a significant 50% and 68% respective increase in Kv4.3 and Kv4.2 SUMOylation relative to PBS. Bath application of the catalytically inactive TAT-PIAS3-ΔSIM1 had no effect, indicating that PIAS3 is a bona fide SUMO E3 ligase for cardiomyocyte Kv4 channels. The experiment was repeated except cells received a 30 min bath application of PBS, 8-bromo-cAMP, or 8-bromo-cAMP + TAT-PIAS3 (Fig. 9F-G). Application of 8-bromo-cAMP reduced but did not significantly alter mean normalized SUMOylation indices. However, co-application of cAMP blocked the previously observed PIAS3-induced increase in channel SUMOylation for both Kv4.3 and Kv4.2. Together, these data indicate that PIAS3 is a SUMO E3 ligase for Kv4 channels in cardiomyocytes, and that PIAS3-mediated SUMOylation can be blocked by an increase in the PKA activator, cAMP.

PIAS3 is a bona fide SUMO E3 ligase for cardiomyocyte Kv4 channels, and PIAS3-mediated SUMOylation of Kv4.2 and Kv4.3 channels is regulated by cAMP levels. A-B Representative results for IP followed by western blot experiments using protein lysates from mouse heart. IP and western blot (WB) antibodies were as indicated. Western blots were first probed with anti-SUMO (A, left) or anti-PIAS3 (B, left), imaged, stripped, and re-probed with anti-Kv4.2 (A&B, right). The experiment was repeated on 3 different heart lysates with the same result. Red asterisks mark Kv4.2. Blue asterisk marks PIAS3. The data show that Kv4.2 channels are SUMOylated and that they interact with PIAS3 in mouse heart. PIAS3 is likely to be post-translationally modified as the signal is routinely higher than the expected molecular weight of ~ 64–69 kD. C Top panel: Micrograph showing SUMOylation of endogenous Kv4 channels in cultured rat neonatal cardiomyocytes (numbered 1–5). Large bright signals (arrowheads) represent noise that was excluded from analyses based on particle size. Bottom panel: Black puncta represent the masks produced by the ImageJ particle analysis program for the 5 cells in the top panel. The 5 cells in the top panel were outlined in yellow and the outlines were overlayed onto the mask to illustrate the RID for all puncta in each cell. The numbers in the bottom panel represent the normalized SUMOylation index for each cell. D-G Tukey box plots derived from normalized SUMOylation indices for each treatment group. All data points were divided by the mean normalized SUMOylation index for the control group. Treatment groups and the number of cells in each treatment group are indicated below the graphs. Panel D: *, significantly different from PBS and TAT-PIAS3-ΔSIM1 treatment groups; Kruskal-Wallis test followed by a Dunn’s multiple comparisons post hoc test that makes all pairwise comparisons, H = 37.59, p < 0.0001; panel E: *, significantly different from PBS and TAT-PIAS3-ΔSIM1 treatment groups; Kruskal-Wallis test followed by a Dunn’s multiple comparisons post hoc test that makes all pairwise comparisons, H = 38.9, p < 0.0001; panel F: Kruskal-Wallis test, H = 2.658, p = 0.2648; panel G: Kruskal-Wallis test, H = 0.02656, p = 0.9868
TC components HA-KChIP2a and HA-DPP10 are not substrates for PIAS3
PIAS3 is known to interact with Kv β-subunits and alter interactions between Kv α- and β-subunits [20, 21]. The Kv4.2g TC comprises pore-forming and auxiliary subunits. The TC will form in the presence or absence of PIAS3; however, E3 SUMO ligases recruited to protein complexes often induce en masse SUMOylation. The process whereby many proteins in a complex are simultaneously SUMOylated is referred to as group SUMOylation [9, 10, 45, 46]. We asked if HA-KChIP2a and HA-DPP10c were targets for PIAS3 when they were incorporated into the TC. HEK cells were transiently transfected with plasmids expressing the Kv4.2g TC with and without PIAS3. Cells were lysed and protein lysates were used in IPs with anti-Kv4.2 followed by western blot experiments.
Figure 10 and Supplemental Fig. 2 show that HA-KChIP2a is decorated by SUMO1 and SUMO2 in the protein lysate. However, only non-SUMOylated HA-KChIP2a proteins were detected in the TC. HA-KChIP2a proteins retrieved from the supernatant fraction of the IP (protein not bound to Kv4.2g) but not the eluant fraction (bound to Kv4.2g) were decorated by SUMO1 and SUMO2 (Fig. 10A, B). Co-expression of PIAS3 had no effect on HA-KChIP2a SUMOylation in the TC. These data suggest that HA-KChIP2a in the TC is not a PIAS3 substrate and that perhaps HA-KChIP2a SUMOylation may prevent incorporation into the TC, though more experimentation is necessary to support this conclusion.

PIAS3 does not alter HA-KChIP2a and HA-DPP10c SUMOylation in the ternary complex. A-B Representative results for IP followed by western blot experiments using protein lysates from HEK cells expressing Kv4.2g TC with or without PIAS3. IP and western blot antibodies were as indicated. Western blots were first probed with anti-SUMO, imaged, stripped, and re-probed with anti-panKChIP. Fractions are indicated below the blot: supernatant, the fraction not bound to Kv4.2g; Kv4.2 IP, the fraction bound to Kv4.2g. Experiments (transfection + IP/western blot) were repeated with the same result, n = 4 per treatment group. C-D Representative results for IP followed by western blot experiment using protein lysates from HEK cells expressing Kv4.2g TC with or without PIAS3. IP and western blot antibodies were as indicated. Western blots were first probed with anti-SUMO, imaged, stripped, and re-probed with anti-HA. E-F Bar graphs show the mean normalized fraction of SUMO conjugated HA-DPP10c ± SEM. The fraction of SUMOylated DPP10c in each experiment was measured as SUMO OD ÷ HA OD. Each data point was then divided by the mean for the control. Each symbol represents one experiment (transfection + IP/western blot). ns, non-significant, (SUMO1: 1.00 ± 0.23 vs. 0.81 ± 0.14, t-test, p = 0.523; SUMO2: 1.00 ± 0.11 vs. 0.93 ± 0.15, t-test, p = 0.706)
Next, SUMOylation was examined for HA-DPP10c incorporated into the TC (Fig. 10C-F). HA-DPP10c proteins were SUMOylated in all fractions (lysate, supernatant, IP). HA-DPP10c comprised by the TC was decorated by SUMO1 (Fig. 10C) and SUMO2 (Fig. 10D). SUMOylation of HA-DPP10c incorporated into the TC was not significantly altered by PIAS3 overexpression (Fig. 10E-F). These data confirm that Kv4.2-K579 is the only substrate for PIAS3 in the TC.
HCN2-K669 is a substrate for PIAS3
Early work on KChAP showed that co-expression of PIAS3 increased the surface expression of additional α-subunits, including Kv1.3 and Kv2.1 [20, 22]. SUMOylation of HCN2-K669 can also increase rab11a-dependent recycling of endocytosed channels resulting in an increase in HCN2 surface expression and the maximal conductance of the hyperpolarization activated current mediated by the channels (Ih) [28, 32]. To determine if PIAS3 could also serve as a SUMO E3 ligase for HCN2 channels, a previously described HEK cell line stably expressing HCN2 (HEK-HCN2) [32] was transiently transfected with mCherry or mCherry + PIAS3, and Ih was recorded with or without SUMO in the patch pipet. PIAS3 expression increased Ih Gmax by ~ 60% compared to control and occluded the effects of increasing SUMO (Fig. 11A-B). To determine if PIAS3 targets K669, experiments were repeated with the previously described SUMO deficient HEK-HCN2-K669R stable line. Ih Gmax was not significantly different between mCherry or mCherry + PIAS3 treatment groups (Fig. 11A&D). These data suggest that HCN2-K669 is also a substrate for PIAS3.

PIAS3 acts at HCN2-K669 to increase IhGmax. A HEK cells stably expressing HCN2 or the SUMOylation deficient HCN2-K669R were or were not transiently transfected with PIAS3. After 48 h, cells were used in whole cell patch clamp experiments to measure Ih Gmax with or without SUMO peptides (4.2 µM) in the recording pipet. The voltage protocol used to elicit the current is shown in the bottom right panel. All other panels show representative current traces for each treatment group. B Bar graphs show mean Ih Gmax ± SEM for cells expressing HCN2. Each symbol is one cell. All data were derived from ≥ 3 replicate experiments for each treatment group. *, significantly different as determined with a one-way ANOVA followed by a Tukey’s post hoc test that makes all pairwise comparisons (F (2,25) = 7.01, p = 0.0038); NS, not significant. C Bar graphs show mean Ih Gmax ± SEM for cells expressing the SUMOylation-deficient HCN2-K669R. Each symbol is one cell. All data were derived from ≥ 3 replicate experiments for each treatment group. The mutation blocked the PIAS3-mediated increase in Ih Gmax (8.44. ± 1.00nS vs. 6.92 ± 0.83nS, t-test, p = 0.263)
Conclusions
Dynamic ion channel SUMOylation shapes the electrical properties of excitable cells. It is mediated by two classes of enzymes with opposing actions. Ion channel deSUMOylation by SENPs is well characterized. This study provides the first information on an E3 ligase that promotes ion channel SUMOylation. PIAS3 belongs to the SP-RING family of SUMO E3 ligases [12]. It shuttles between the nucleus and cytoplasm [47]. PIAS3 catalyzed SUMOylation at Kv4.2-K579 and HCN2-K669, which increased channel conductances. PKA-mediated phosphorylation at Kv4.2-S552 prevented SUMOylation at Kv4.2-K579 and blocked the PIAS3 mediated increase in IA Gmax. This suggests that modulatory tone may shape ion channel surface expression through post-translational modification crosstalk.
PIAS3-mediated C-terminal SUMOylation increases ion channel surface expression
We previously showed that overexpression of SUMO2 + ubc9 enhanced SUMOylation at Kv4.2-K579, which increased IA Gmax mediated by a Kv4.2g ternary complex in HEK cells [27]. Western blot experiments showed Kv4.2-K579 SUMOylation did not alter channel stability. Internalization and biotinylation assays demonstrated that Kv4.2-K579 SUMOylation decreased channel internalization, resulting in increased surface expression. A 20 min blockade of clathrin-mediated endocytosis mimicked and occluded the effect of enhanced Kv4.2-K579 SUMOylation on IA Gmax, confirming that SUMOylation reduced channel internalization. Internalization depends on two opposing processes: endocytosis and recycling. A 20 min blockade of clathrin-mediated endocytosis will disrupt both processes. Additional experiments showed that rab11a, which regulates channel recycling, was necessary for the SUMOylation induced increase in IA Gmax [28]. Co-IP experiments showed that the interaction between Kv4.2 and the α-subunit of the AP-2 complex, which is responsible for clathrin-mediated endocytosis, was unchanged by SUMOylation [27]. In sum, Kv4.2-K579 SUMOylation increased IA Gmax by reducing channel internalization, thereby increasing channel surface expression. Reduced internalization was due, at least in part, to an increase in channel recycling. Additional experiments are necessary to determine if Kv4.2-K579 SUMOylation also reduced channel endocytosis.
This study provides compelling evidence that PIAS3 acts by catalyzing Kv4.2-K579 SUMOylation. First, like effects were produced by overexpressing PIAS3 or SUMO + ubc9; they both enhanced channel SUMOylation and increased IA Gmax in HEK cells through a rab11a-dependent mechanism when IA was mediated by wild type α-subunits (Kv4.2-K579), but not when IA was mediated by SUMOylation-deficient α-subunits (Kv4.2-K579R). Second, the SUMO-conjugating enzyme, ubc9, was necessary for PIAS3 effects on IA Gmax in HEK cells. Third, the PIAS3 SIM1 domain, which orients the donor SUMO for conjugation to the target protein [12, 17], was necessary for PIAS3 effects on IA Gmax in HEK cells. Fourth, PLA experiments showed that wild type PIAS3, but not a catalytically inactive PIAS3, increased SUMOylation of endogenous Kv4.2 and Kv4.3 channels in cardiomyocytes. The data presented here, together with our previous work, suggest that PIAS3 catalyzes Kv4.2-K579 SUMOylation, which results in enhanced channel recycling, increased cannel surface expression and a rise in IA Gmax.
Previous work showed that PIAS3 could regulate several ion channels [20, 22]. Here the list was expanded to include Kv4.2 and HCN2 channels. Those channels whose surface expression increased in response to enhanced PIAS3 activity (i.e., Kv1.3, Kv2.1/2, Kv4.2/3, and HCN2) all have distal C-terminal SUMOylation sites. PIAS3 could potentially co-regulate the recycling of a battery of ion channels by controlling their C-terminal SUMOylation status.
Rab-11a dependent recycling was necessary for the SUMOylation-induced increase in HCN2 and Kv4.2 surface expression in HEK cells (Figs. 3 and [28]). HCN2 [48] and Kv4.2 [49] channels co-localize with the perinuclear endocytic recycling compartment (ERC) in opossum kidney cells and HEK293 cells, respectively. SUMOylation enhanced HCN2 co-localization with the ERC in HEK293 cells [28]; Kv4.2 was not tested. In this study, SUMOylated Kv4.2 and Kv4.3 channels also displayed a predominately perinuclear localization in cardiomyocytes. An important unanswered question is: Does Kv4 SUMOylation regulate channel recycling in cardiomyocytes?
Crosstalk between post-translational modifications
The phosphorylation status of a target protein can play a permissive role in target protein SUMOylation; phosphorylation can either promote or prevent SUMOylation [46, 50]. A conserved 68 amino acid fragment (S552-S620) contains the Kv4.2-K579 SUMOylation site surrounded by 10 phosphorylation sites [42]. This could represent a regulatory module that integrates parallel signals to influence a binary output (K vs. K + SUMO) that controls channel surface expression. Consistent with this hypothesis, western blot experiments with a phosphomimetic Kv4.2 construct showed that mimicking phosphorylation at Kv4.2-S552 blocked SUMOylation at Kv4.2-K579 in HEK cells co-expressing HA-KChIP2a + HA-DPP10c (Fig. 8). The most parsimonious interpretation of the data is that PKA-dependent phosphorylation impairs PIAS3-mediated re-insertion of Kv4.2 into the plasma membrane, contributing to the overall reduction in surface Kv4.2.
The effect of phosphorylation has been studied for 4 of the 10 sites in the putative regulatory module. One important caveat to keep in mind is that the effect of a post-translational modification is context dependent. The consequences of Kv4.2-S552 phosphorylation are reliant upon the subunit composition of the channel complex and the stage at which phosphorylation occurs during channel assembly and trafficking [51,52,53]. Similarly, the effect of Kv4.2-K579 SUMOylation depends upon the subunit composition of the channel complex [26, 27].
Heterologous expression studies in Xenopus oocytes showed channel biophysical properties were uniquely altered by MAP kinase phosphorylation of Kv4.2-T602, Kv4.2-T607, and Kv4.2-S616 [52, 54]. MAP kinases also decreased Kv4.2 channel surface expression in HEK293 cells and hippocampal neurons, but the specific phosphorylation site(s) was not identified [49]. Elegant experiments showed that MAP kinases decreased Kv4.2 channel endocytosis in both cell types, but the effect of MAP kinases on channel recycling was not examined. The SUMOylation status of Kv4.2-K579 was not considered in any previous MAP kinase study, and it is not known if these sites play a permissive role in Kv4.2-K579 SUMOylation.
PKA can phosphorylate Kv4.2-S552 to increase channel internalization in heterologous expression systems, neurons, and cardiomyocytes [44, 55, 56]. PKA-induced internalization was blocked by inhibiting clathrin-mediated endocytosis for ≥ 10 min [43], which has the potential to disrupt both endocytosis and recycling; thus, the mechanism(s) by which PKA increases internalization in previous studies remains unresolved. In our experiments where both PKA and PIAS3 activities were globally elevated in HEK cells and cardiomyocytes, post-translational modification crosstalk occurred and phosphorylation blocked SUMOylation; however, under physiological conditions the effect of a post-translational modification may be spatially and/or temporally constrained, and the same level of crosstalk may not occur. It will be important to determine where the post-translational modification crosstalk takes place. PKA is tethered to membranes of all subcellular compartments that are active in endocytosis and recycling [57, 58]. Colocalization of the SUMOylation machinery with membranes is not well documented. Applying SUMO to the intracellular face of excised membrane patches stably increases channel SUMOylation [6], indicating that ubc9 is tightly associated with the plasma membrane and SUMOylation can occur there. It is not clear if PIAS3 is also associated with the plasma membrane or if either enzyme is associated with intracellular membranes involved in recycling.
Post-translational modification crosstalk may contribute to signaling in excitable cells
This study showed that Kv4.2 channels were SUMOylated in cardiomyocytes, that PIAS3 enhanced their SUMOylation, and increasing cytosolic cAMP blocked the PIAS3-mediated increase in Kv4.2/3 SUMOylation. In rodent cardiomyocytes, Kv4.2 and Kv4.3 heterotetramers mediate Itof [59], which controls initial repolarization of the action potential and Ca2+ entry [60, 61]. PKA is a negative regulator of Itof. Stimulation of cardiomyocyte α1-adrenergic receptors produced a PKA-mediated reduction in Kv4.2 surface expression and Itof, [62, 63], but only for K4.2 channels localized to caveolae lipid rafts [56, 64, 65]. Itof is also reduced during acute ischemia by the β-adrenergic receptor-PKA axis [66]. It will be interesting to determine if Kv4.2-K579 and Kv4.3-K577 SUMOylation are necessary for the effects of adrenergic signaling.
Post-translational modification crosstalk could also play a role in neurons. Kv4.2 channels are located in dendrites of hippocampal neurons where they modulate action potential back propagation and synaptic integration [67, 68]. PIAS3 is localized to pre- and post-synaptic compartments in the dendrites of hippocampal neurons [69], and many synaptic proteins are SUMOylated [2]. Internalization of hippocampal Kv4.2 channels is increased by PKA-mediated phosphorylation of Kv4.2-S552 during LTP [43, 44], but SUMO involvement has never been investigated. PKA regulation of Kv4 SUMOylation may contribute to activity-dependent homeostasis involving IA and Ih. Studies on an identified invertebrate neuron, LP, suggested that dopamine acted through the D1 receptor-PKA axis to gate activity dependent SUMOylation that maintained LP duty cycle. In this case, tonic nM dopamine permitted and prevented short-term activity-dependent regulation of LP Ih and IA, respectively [70, 71].
In sum, PIAS3 is an important, understudied regulator of ion channels. It influences the surface expression of at least four classes of voltage-gated α-subunits, and it can regulate interactions between Kv α- and β-subunits. Cross talk between PKA-mediated phosphorylation and PIAS3-mediated SUMOylation may play a key role in remodeling electrical activity in excitable cells.
Availability of data and materials
The dataset(s) supporting the conclusions of this article is(are) included within the article.
Data availability
No datasets were generated or analysed during the current study.
References
Silveirinha VC, et al. CaV2.2 (N-type) voltage-gated calcium channels are activated by SUMOylation pathways. Cell Calcium. 2021;93: 102326. https://doi.org/10.1016/j.ceca.2020.102326.
Henley JM, et al. SUMOylation of synaptic and synapse-associated proteins: an update. J Neurochem. 2021;156(2):145–61. https://doi.org/10.1111/jnc.15103.
Kotler O, et al. SUMOylation of na(V)1.2 channels regulates the velocity of backpropagating action potentials in cortical pyramidal neurons. Elife. 2023;12. https://doi.org/10.7554/eLife.81463.PMC10014073.
Lv YY, et al. SUMOylation of Kir7.1 participates in neuropathic pain through regulating its membrane expression in spinal cord neurons. CNS Neurosci Ther. 2022;28(8):1259–67. https://doi.org/10.1111/cns.13871.PMC9253747.
Xu Y, et al. Hypoxia inhibits the cardiac I K1 current through SUMO targeting Kir2.1 activation by PIP2. iScience. 2022;25(9):104969. https://doi.org/10.1016/j.isci.2022.104969.PMC9437851.
Plant LD, et al. Hypoxia produces pro-arrhythmic late sodium current in cardiac myocytes by SUMOylation of NaV1.5 channels. Cell Rep. 2020;30(7):2225-2236 e4. https://doi.org/10.1016/j.celrep.2020.01.025.PMC7054841.
Xiong D, et al. SUMOylation determines the voltage required to activate cardiac IKs channels. Proc Natl Acad Sci U S A. 2017;114(32):E6686-94. https://doi.org/10.1073/pnas.1706267114.PMC5559042.
Hasan R, et al. SUMO1 modification of PKD2 channels regulates arterial contractility. Proc Natl Acad Sci U S A. 2019;116(52):27095–104. https://doi.org/10.1073/pnas.1917264116.PMC6936352.
Vertegaal ACO. Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol. 2022. https://doi.org/10.1038/s41580-022-00500-y.
Celen AB, Sahin U. Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts. FEBS J. 2020;287(15):3110–40. https://doi.org/10.1111/febs.15319.
Hendriks IA, et al. System-wide identification of wild-type SUMO-2 conjugation sites. Nat Commun. 2015;6:7289. https://doi.org/10.1038/ncomms8289.4490555.
Varejao N, et al. Molecular mechanisms in SUMO conjugation. Biochem Soc Trans. 2020;48(1):123–35. https://doi.org/10.1042/BST20190357.
Hotz PW, Muller S, Mendler L. SUMO-specific isopeptidases tuning cardiac SUMOylation in health and disease. Front Mol Biosci. 2021;8:786136. https://doi.org/10.3389/fmolb.2021.786136.PMC8641784.
Chen X, et al. The SUMO-specific protease SENP2 plays an essential role in the regulation of Kv7.2 and Kv7.3 potassium channels. J Biol Chem. 2021;297(4):p101183. https://doi.org/10.1016/j.jbc.2021.101183. PMC8488601.
Gomez K, et al. Targeted transcriptional upregulation of SENP1 by CRISPR activation enhances deSUMOylation pathways to elicit antinociception in the spinal nerve ligation model of neuropathic pain. Pain. 2023. https://doi.org/10.1097/j.pain.0000000000003080.
Nishida T, Yasuda H. PIAS1 and PIASxalpha function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J Biol Chem. 2002;277(44):41311–7. https://doi.org/10.1074/jbc.M206741200.
Lascorz J, et al. SUMO-SIM interactions: from structure to biological functions. Semin Cell Dev Biol. 2022;132:193–202. https://doi.org/10.1016/j.semcdb.2021.11.007.
Lussier-Price M, et al. Characterization of a C-terminal SUMO-interacting motif present in select PIAS-family proteins. Structure. 2020;28(5):573-585 e5. https://doi.org/10.1016/j.str.2020.04.002.
Varejao N, et al. Structural basis for the E3 ligase activity enhancement of yeast Nse2 by SUMO-interacting motifs. Nat Commun. 2021;12(1):7013. https://doi.org/10.1038/s41467-021-27301-9.PMC8636563.
Wible BA, et al. Cloning and expression of a novel K + channel regulatory protein, KChAP. J Biol Chem. 1998;273(19):11745–51. https://doi.org/10.1074/jbc.273.19.11745.
Kuryshev YA, et al. KChAP/Kvbeta1.2 interactions and their effects on cardiac Kv channel expression. Am J Physiol Cell Physiol. 2001;281(1):C290-9. https://doi.org/10.1152/ajpcell.2001.281.1.C290.
Kuryshev YA, et al. KChAP as a chaperone for specific K(+) channels. Am J Physiol Cell Physiol. 2000;278(5):C931-41. https://doi.org/10.1152/ajpcell.2000.278.5.C931.
Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell. 2001;8(3):713–8. https://doi.org/10.1016/s1097-2765(01)00349-5.
Johnson ES, Gupta AA. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell. 2001;106(6):735–44. https://doi.org/10.1016/s0092-8674(01)00491-3.
Martin S, et al. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447(7142):321–5. https://doi.org/10.1038/nature05736.3310901.
Welch MA, et al. SUMOylating two distinct sites on the A-type potassium channel, Kv4.2, increases surface expression and decreases current amplitude. Front Mol Neurosci. 2019;12(144): 144. https://doi.org/10.3389/fnmol.2019.00144.
Welch MA, Jansen LR, Baro DJ. SUMOylation of the Kv4.2 ternary complex increases surface expression and current amplitude by reducing internalization in HEK 293 cells. Front Mol Neurosci. 2021;14:757278. https://doi.org/10.3389/fnmol.2021.757278.PMC8593141.
Forster LA, Baro DJ. C-terminal post-translational modification of ion channels by the small ubiquitin like modifier (SUMO) promotes rab11a mediated slow recycling of endocytosed channels. Program No. 189.06. 2022 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience; 2022. Online.
Zu S, et al. Duck PIAS2 negatively regulates RIG-I mediated IFN-beta production by interacting with IRF7. Dev Comp Immunol. 2020;108:103664. https://doi.org/10.1016/j.dci.2020.103664.
Onishi S, Kataoka K. PIASy is a SUMOylation-independent negative regulator of the insulin transactivator MafA. J Mol Endocrinol. 2019;63(4):297–308. https://doi.org/10.1530/JME-19-0172.
Li R, et al. PIAS1 negatively modulates virus triggered type I IFN signaling by blocking the DNA binding activity of IRF3. Antiviral Res. 2013;100(2):546–54. https://doi.org/10.1016/j.antiviral.2013.09.001.
Parker AR, et al. SUMOylation of the hyperpolarization-activated cyclic nucleotide-gated channel 2 increases surface expression and the maximal conductance of the hyperpolarization-activated current. Front Mol Neurosci. 2017;9:168. https://doi.org/10.3389/fnmol.2016.00168.5226956.
Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569(Pt 1):41–57. https://doi.org/10.1113/jphysiol.2005.095042.PMC1464206.
Chung CD, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278(5344):1803–5. https://doi.org/10.1126/science.278.5344.1803.
Baro DJ, et al. Alternate splicing of the shal gene and the origin of I(A) diversity among neurons in a dynamic motor network. Neuroscience. 2001;106(2):419–32.
Kim M, et al. Expression of Panulirus shaker potassium channel splice variants. Receptors Channels. 1998;5(5):291–304.
Allgood SC, Neunuebel MR. The recycling endosome and bacterial pathogens. Cell Microbiol. 2018;20(7):e12857. https://doi.org/10.1111/cmi.12857.PMC5993623.
Ren M, et al. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc Natl Acad Sci U S A. 1998;95(11):6187–92. https://doi.org/10.1073/pnas.95.11.6187.PMC27621.
Walrant A, et al. Membrane crossing and membranotropic activity of cell-penetrating peptides: dangerous liaisons? Acc Chem Res. 2017;50(12):2968–75. https://doi.org/10.1021/acs.accounts.7b00455.
Gonzalez-Prieto R, et al. Global non-covalent SUMO interaction networks reveal SUMO-dependent stabilization of the non-homologous end joining complex. Cell Rep. 2021;34(4): 108691. https://doi.org/10.1016/j.celrep.2021.108691.
Namanja AT, et al. Insights into high affinity small ubiquitin-like modifier (SUMO) recognition by SUMO-interacting motifs (SIMs) revealed by a combination of NMR and peptide array analysis. J Biol Chem. 2012;287(5):3231–40. https://doi.org/10.1074/jbc.M111.293118.PMC3270977.
Hu JH, Liu Y, Hoffman DA. Identification of Kv4.2 protein complex and modifications by tandem affinity purification-mass spectrometry in primary neurons. Front Cell Neurosci. 2022;16:1070305. https://doi.org/10.3389/fncel.2022.1070305.PMC9788671.
Kim J, et al. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K + channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54(6):933–47. https://doi.org/10.1016/j.neuron.2007.05.026.1950443.
Hammond RS, et al. Protein kinase A mediates activity-dependent Kv4.2 channel trafficking. J Neurosci. 2008;28(30):7513–9. https://doi.org/10.1523/JNEUROSCI.1951-08.2008.2665045.
Psakhye I, Jentsch S. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell. 2012;151(4):807–20. https://doi.org/10.1016/j.cell.2012.10.021.
Flotho A. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. 2013;82:357–85. https://doi.org/10.1146/annurev-biochem-061909-093311.
Thirouin ZS, et al. Trophic factor BDNF inhibits GABAergic signaling by facilitating dendritic enrichment of SUMO E3 ligase PIAS3 and altering gephyrin scaffold. J Biol Chem. 2022;298(5):101840. https://doi.org/10.1016/j.jbc.2022.101840.PMC9019257.
Hardel N, et al. Recycling endosomes supply cardiac pacemaker channels for regulated surface expression. Cardiovasc Res. 2008;79(1):52–60. https://doi.org/10.1093/cvr/cvn062.
Tabor GT, et al. A novel bungarotoxin binding site-tagged construct reveals MAPK-dependent Kv4.2 trafficking. Mol Cell Neurosci. 2019;98:121–30. https://doi.org/10.1016/j.mcn.2019.06.007.PMC6639039.
Hendriks IA, et al. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol. 2017;24(3):325–36. https://doi.org/10.1038/nsmb.3366.
Schrader LA, et al. PKA modulation of Kv4.2-encoded A-type potassium channels requires formation of a supramolecular complex. J Neurosci. 2002;22(23):10123–33.
Schrader LA, et al. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol. 2006;290(3):C852-61. https://doi.org/10.1152/ajpcell.00358.2005.
Lin L, et al. KChIP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol Cell Neurosci. 2010;43(3):315–25. https://doi.org/10.1016/j.mcn.2009.12.005.2823810.
Adams JP, et al. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75(6):2277–87. https://doi.org/10.1046/j.1471-4159.2000.0752277.x.
Anderson AE, et al. Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. J Biol Chem. 2000;275(8):5337–46.
Li Y, et al. Kv4.2 phosphorylation by PKA drives Kv4.2-KChIP2 dissociation, leading to Kv4.2 out of lipid rafts and internalization. Am J Physiol Cell Physiol. 2022;323(1):C190-C. https://doi.org/10.1152/ajpcell.00307.2021.
Griffiths G, et al. Ultrastructural localization of the regulatory (RII) subunit of cyclic AMP-dependent protein kinase to subcellular compartments active in endocytosis and recycling of membrane receptors. J Cell Sci. 1990;96(Pt 4):691–703. https://doi.org/10.1242/jcs.96.4.691.
Lin L, et al. AKAP79/150 impacts intrinsic excitability of hippocampal neurons through phospho-regulation of A-type K + channel trafficking. J Neurosci. 2011;31(4):1323–32. https://doi.org/10.1523/JNEUROSCI.5383-10.2011.3035425.
Nerbonne JM. Molecular basis of functional myocardial potassium channel diversity. Card Electrophysiol Clin. 2016;8(2):257–73. https://doi.org/10.1016/j.ccep.2016.01.001. PMC4893780.
Johnson EK, et al. Differential expression and remodeling of transient outward potassium currents in human left ventricles. Circ Arrhythm Electrophysiol. 2018;11(1):e005914. https://doi.org/10.1161/CIRCEP.117.005914.PMC5775893.
Marionneau C, et al. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K + currents with left ventricular hypertrophy. Circ Res. 2008;102(11):1406–15. https://doi.org/10.1161/CIRCRESAHA.107.170050.PMC2653713.
Gallego M, et al. Alpha1-adrenoceptors stimulate a Galphas protein and reduce the transient outward K + current via a cAMP/PKA-mediated pathway in the rat heart. Am J Physiol Cell Physiol. 2005;288(3):C577-85. https://doi.org/10.1152/ajpcell.00124.2004.
Gallego M, Casis O. Regulation of cardiac transient outward potassium current by norepinephrine in normal and diabetic rats. Diabetes Metab Res Rev. 2001;17(4):304–9. https://doi.org/10.1002/dmrr.212.
Alday A, et al. Alpha1-adrenoceptors regulate only the caveolae-located subpopulation of cardiac K(V)4 channels. Channels (Austin). 2010;4(3):168–78. https://doi.org/10.4161/chan.4.3.11479.
Gallego M, et al. Adrenergic regulation of cardiac ionic channels: role of membrane microdomains in the regulation of kv4 channels. Biochim Biophys Acta. 2014;1838(2):692–9. https://doi.org/10.1016/j.bbamem.2013.06.025.
Zhang L, et al. Propranolol regulates cardiac transient outward potassium channel in rat myocardium via cAMP/PKA after short-term but not after long-term ischemia. Naunyn Schmiedebergs Arch Pharmacol. 2010;382(1):63–71. https://doi.org/10.1007/s00210-010-0520-y.
Hoffman DA, et al. K + channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387(6636):869–75. https://doi.org/10.1038/43119.
Ramakers GM, Storm JF. A postsynaptic transient K(+) current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 2002;99(15):10144–9. https://doi.org/10.1073/pnas.152620399.PMC126638.
Conz A, et al. Super-resolution study of PIAS SUMO E3-ligases in hippocampal and cortical neurons. Eur J Histochem. 2021;65(s1). https://doi.org/10.4081/ejh.2021.3241.PMC8419632.
Parker AR, Forster LA, Baro DJ. Modulator-gated, SUMOylation-mediated, activity-dependent regulation of ionic current densities contributes to short-term activity homeostasis. J Neurosci. 2019;39(4):596–611. https://doi.org/10.1523/JNEUROSCI.1379-18.2018.PMC6343643.
Krenz WD, Rodgers EW, Baro DJ. Tonic 5nM DA stabilizes neuronal output by enabling bidirectional activity-dependent regulation of the hyperpolarization activated current via PKA and calcineurin. PLoS One. 2015;10(2):e0117965. https://doi.org/10.1371/journal.pone.0117965.4333293.
Acknowledgements
The authors thank Dr. Ruth Harris for the use of her equipment. We also thank Micheal Kyere and Leah Middleton for excellent technical assistance. In addition, we thank Kirin Gada for sharing her expertise on cardiomyocyte culturing and recording.
Funding
D.J.B. acknowledges support from NIH R03 NS116327 and seed grants from the GSU MBD and B&B programs. L-ARJ and MAW were GSU B&B Fellows.
Author information
Authors and Affiliations
Contributions
LRJ, MAW and DJB performed experiments and analyzed data. All authors contributed to the conception and design of the work. LRJ and DJB wrote the manuscript. All authors read, edited and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12964_2024_1795_MOESM1_ESM.tif
Supplementary Material 1. Supplemental Figure 1. Measurements of Kv4.2 SUMOylation. Uncropped blots showing typical results for Kv4.2 (A) and Kv4.2-K579 (B) in IP/western blot experiments to measure Kv4.2 protein SUMOylation. Asterisks on the left indicate the signal representing the ~65kD Kv4.2 proteins (no GFP tag) from the IP experiments. Asterisks on the right indicate Kv4.2 proteins in the lysate. The Kv4.2 proteins in each preparation run to discrete positions, despite being the same size, most likely due to differences in the buffer composition for each preparation and perhaps protein concentrations. Additional bands on the SUMO1 blot that are not present on the Kv4.2 blot represent SUMOylated proteins that co-IP with Kv4.2, such as HA-DPP10c (~100kD). Bands above the asterisks that are present on the Kv4.2 and SUMO1 blots represent post-translationally modified and/or aggregated Kv4.2 proteins. The only IP signals quantified were those indicated by the asterisk. Note 20mL of IP product or 10mg of protein lysate was run in each lane. The loss of Kv4.2 signal intensity in the lysate lanes on the Kv4.2 blot relative to the SUMO1 blot is partially due to protein loss as a result of stripping of the blot before probing for Kv4.2. In addition, the SUMO1 signal in the lysate lanes represents the entire SUMOylome (above 50kD).
12964_2024_1795_MOESM2_ESM.tif
Supplementary Material 2. Supplemental Figure 2. Typical results from experiments to measure DPP10c and KChIP2a SUMOylation. Uncropped blots showing typical results from IP followed by western blot experiments to measure SUMOylation of DPP10c (A) and KChIP2a (B). Asterisks indicate the protein of interest that was measured: A. HA-DPP10c, ~100kD; B. KChIP2a, ~41kD. The different fractions from the IP experiments are indicated by E for eluant (bound to anti-Kv4.2), S for supernatant (not bound to anti-Kv4.2), and L for lysate used in IP experiments. Only the bands indicated by the asterisk were quantified. Note 20mL of IP product or 10mg of protein lysate was run in each lane.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jansen, LA.R., Welch, M.A., Plant, L.D. et al. Crosstalk between PKA and PIAS3 regulates cardiac Kv4 channel SUMOylation. Cell Commun Signal 22, 422 (2024). https://doi.org/10.1186/s12964-024-01795-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01795-4