
Abstract
Oncolytic viruses (OVs) for cancer treatment are in a rapid stage of development, and the direct tumor lysis and activation of a comprehensive host immune response are irreplaceable advantages of cancer immunotherapy. However, excessive antiviral immune responses also restrict the spread of OVs in vivo and the infection of tumor cells. Macrophages are functionally diverse innate immune cells that phagocytose tumor cells and present antigens to activate the immune response, while also limiting the delivery of OVs to tumors. Studies have shown that the functional propensity of macrophages between OVs and tumor cells affects the overall therapeutic effect of oncolytic virotherapy. How to effectively avoid the restrictive effect of macrophages on OVs and reshape the function of tumor-associated macrophages in oncolytic virotherapy is an important challenge we are now facing. Here, we review and summarize the complex dual role of macrophages in oncolytic virotherapy, highlighting how the functional characteristics of macrophage plasticity can be utilized to cooperate with OVs to enhance anti-tumor effects, as well as highlighting the importance of designing and optimizing delivery modalities for OVs in the future.
Similar content being viewed by others

Introduction
Tumors often acquire immunosuppressive mechanisms through cancer immunoediting, which shapes the tumor microenvironment (TME) for tumor growth, effectively avoiding immune-mediated tumor clearance [1, 2]. In fact, cells in the TME are both tumor-supportive and tumor-suppressive, and the function of these cells is influenced by the type of cancer, the individual development of the TME cells, and the degree of their “education” [3].
Currently, tumor immunotherapy has received increasing attention with the focus on reducing tumor-associated immune evasion as the main therapeutic strategy for cancer treatment and depleting or “re-educating” cancer-promoting microenvironment cells into an immune-stimulating phenotype [3, 4], which serves as an effective means of cancer treatment by interfering with the body's immune response, and has triggered a revolution in the field of oncology [5, 6].
Oncolytic virotherapy is an emerging immunotherapy, where oncolytic viruses (OVs) preferentially infect tumor cells to suppress tumor growth and are safe for other normal cells. Above this, OVs can activate the host immune system to generate an immune response to enhance anti-tumor efficacy [7], which can overcome cancer-associated immunosuppression and reshape the tumor microenvironment by increasing the infiltration of immune effector cells in “cold” tumors, making them “hot”. This immune response is becoming a decisive factor in the efficacy of viral therapy [8, 9].
However, since OVs are essentially immunogenic infection factors, they stimulate the host to generate an immune response that can both produce antitumor effects and limit the activity of the virus itself [10]. The delivery of OVs to the host initiates a series of immune responses, and macrophages are one of the professional antigen-presenting cells that bridge innate and adaptive immunity, with major roles in both anti-pathogen infection and anti-tumor immunity [11, 12]. However, the clearance of tumor cells and viruses by macrophages is complex, which is determined by the phenotype and function of macrophages. For one, phagocytosis and antigen presentation by macrophages synergize with OVs to inhibit tumor growth, while OVs also stimulate the shift of macrophages to an antitumor phenotype; as for another, macrophages in turn mediate the clearance of OVs through the secretion of interferon type I (IFN) and the phagocytosis of OVs particles, a role that is an important factor in limiting the systemic administration of OVs [13, 14].
The functional tendency of macrophages between OVs and tumor cells affects the overall therapeutic effect of oncolytic virotherapy, and how to eliminate the detrimental effect of macrophages on OVs and make them work in an anti-tumor direction is an important challenge nowadays. Therefore, in this review, we discuss the essential features and interactions between OVs and macrophages in antitumor immunity, and explore strategies by which to intervene with macrophages in oncolytic virotherapy to improve efficacy.
Oncolytic virotherapy and antitumor immunity
Compared with conventional tumor immunotherapy, oncolytic virotherapy has several advantages, including significant killing effect, high degree of targeting, low adverse effects, and less susceptibility to drug resistance [15, 16]. This is related to the unique antitumor mechanism of OVs, which induces tumor death based on natural interactions among viruses, tumor cells and TME, and the body's immune system (Fig. 1), including direct lysis of tumor cells, promotion of immunogenic cell death (ICD), induction of innate and adaptive immune responses, regulation of TME, and inhibition of tumor angiogenesis [17,18,19]. Furthermore, OVs can be genetically engineered to exert specific effects to enhance anti-tumor functions, the most prevalent of which include tumor targeting, expression of pro-inflammatory cytokines, and tumor suppressor genes [20, 21].

Basic anti-tumor mechanisms of OVs. OVs inhibit tumor growth by selectively infecting and lysing tumor cells, inducing anti-tumor innate and adaptive immunity in the body, and modulating immunosuppressive cells, extracellular matrix cells, and inhibiting tumor angiogenesis in the TME
Direct tumor lysis
In normal host cells, OVs are sensed and cleared by activated antiviral signaling pathways, thus protecting normal cells from OVs damage. However, in tumor cells, overexpression of tumor antigens favorable for OVs binding, availability of a large number of nucleotides, aberrant activation of the oncogenic pathway, dysregulation of the apoptotic pathway, and deficiency of antiviral type I IFN signaling provide OVs with a suitable growth environment and a shelter [22, 23], which allows them to selectively infect and destroy tumor cells, and further releases more progeny of OVs for spreading to neighboring uninfected tumor cells, resulting in an increasing lysis effect [15].
Enhance host immune response
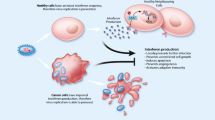
Another important role of OVs is the enhancement of the antitumor immune response through the body-activated antiviral immune response. After delivery of OVs, the immune system preferentially initiates an antiviral response, including the release of antiviral cytokines such as IFNs, tumor necrosis factor-α (TNF-α), and interleukin 12 (IL-12) [24], with these cytokines promoting the maturation of dendritic cells (DCs), which induces a natural antiviral immune response in macrophages and natural killer (NK) cells. Activated NK cells expressing γ-interferon (IFN-γ) and TNF-α contribute further to the activation of macrophages, DCs, and T cells, amplifying the immune response [8, 20], which are recruited to the site of virus-infected tumors, and altering the cytokine environment and the immune cells in the TME, thereby overcoming tumor-associated immunosuppression [25]. Tumor cells invaded and lysed by OVs release progeny viruses, pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), tumor-associated antigens (TAAs), and neoantigens to the extracellular space, and the resulting progeny viruses spread to the surrounding area and infect more tumor cells, creating a tendency for tumor lysis. Whereas DAMPs and PAMPs stimulate the immune system by activating pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) on a variety of immune cells, initiating cellular activation and inflammatory signaling [9]. TAAs and neoantigens are taken up by antigen-presenting cells (APCs) and delivered to T cells, generating viral/tumor antigen-specific CD8+ T cells, which create an immunostimulatory microenvironment at the tumor site, while at the same time, these variations contribute to the transformation of tumor-supportive M2-like macrophages into tumor-suppressive M1-like macrophages [24].
Effect of OVs on TME
The TME provides favorable conditions for tumorigenesis and progression. TME usually includes infiltrative inflammatory cells such as macrophages, lymphocytes, NK cells, and DCs; immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs); cancer-associated fibroblasts (CAFs) tumor stromal cells, and tumor vascular system [3, 26]. Tumor cells co-secrete growth factors, cytokines, and chemokines with immunosuppressive cellular cells that inhibit the normal anti-tumor immune response, provide support for tumor development and angiogenesis, promote tumor progression, and limit therapeutic response [26,27,28,29].
Natural or modified OVs can remodel the immunosuppressive TME, increase the influx of anti-tumor immune cells such as macrophages, NK cells, DCs, T cells, and neutrophils, convert MDSCs into a tumor-killing phenotype, reduce the populations of immune-suppressing cells, remodel the tumor extracellular matrix, and resist tumor angiogenesis, thus promoting tumor elimination by making immunologically “cold” tumors “hot” [30, 31].
In short, OVs can act directly or indirectly on different parts of the tumor ecosystem such as tumor cells, immune cells and cytokines, tumor stromal cells, and tumor vasculature system to inhibit tumor progression.
Macrophages and antitumor immunity
Origin and distribution of macrophages
Macrophages are ubiquitous in any part of the body and perform three essential functions, namely phagocytosis, exogenous antigen presentation, and secretion of cytokines and growth factors for immunomodulation. They perform important duties in tissue development, homeostasis, clearance of dead cells and foreign pathogens, and modulation of inflammatory and tumoral immune responses [32,33,34]. Macrophages also have different names and functions in different tissues, such as circulating monocyte-derived macrophages, tissue-resident macrophages (TRMs), and tumor-associated macrophages, which have complex correlations in terms of classification and origin. TRMs perform appropriate functions in various tissues of the body, including microglia in the brain, Kupffer cells in the liver, and Langerhans cells in the skin [35, 36], and it is currently believed that most of the population of TRMs originates from embryonic precursors in the yolk sac and fetal liver and that they self-maintain independently of the myeloid cells in adulthood [37, 38]. TAMs, on the other hand, consist mainly of circulating monocyte-derived macrophages and RTMs recruited by tumors into TME and are one of the important targets for tumor immunotherapy [39].
Phenotype and function of macrophages
Macrophages are significant plastic and their activation state is influenced by a multitude of factors, but they can usually be simplified into two classifications based on stimulatory factors and secretory products (Fig. 2), namely classically activated M1 macrophages and alternatively activated M2 macrophages [40]. Although this M1/M2 dichotomization simplifies the differences in phenotypic and functional continuum changes in macrophages, this terminology is still more commonly used when discussing whether macrophages are more biased toward a pro-inflammatory or anti-inflammatory phenotype [41].

Macrophage activation and M1/M2 typing. Macrophages polarized into classically activated (M1) or alternatively activated (M2) macrophages under the influence of different cytokines or other factors secrete different cytokines to change the cellular microenvironment to a pro-inflammatory or anti-inflammatory state, exerting anti-tumor or pro-tumor effects at the tumor site
M1 macrophage polarization is usually driven by granulocyte–macrophage colony-stimulating factor (GM-CSF), lipopolysaccharide (LPS), IFN-γ, TNF-α, and PAMPs [42]. M1 phenotype macrophages have mainly pro-inflammatory properties, promoting the pro-inflammatory response of helper T cells 1 (Th1) by secreting cytokines, such as TNF-α, IL-1β, IL-12, and IL-18, and enhancing the recruitment of Th1 cells to sites of inflammation by secreting chemokines, such as chemokines CXC motif ligand 9 (CXCL9) and CXCL10 [43]. M1 macrophages can trigger an adaptive immune response through self-mediated cytotoxicity or cross-presentation of antigens (TAAs and TANs), triggering potent anti-tumor immunity. Therefore, M1 macrophages are considered a tumor-suppressive macrophage phenotype [44].
M2 macrophage polarization is usually driven by macrophage colony-stimulating factor (M-CSF), IL-4, IL-10, IL-13, and transforming growth factor-β (TGF-β) [45]. M2 macrophages have a critical position in appropriate immune function and homeostasis in vivo, with examples including stimulation of Th2 cell responses, mediation of parasite clearance, immunomodulation, wound healing and tissue repair [46]. However, the function of M2 macrophages can also be adversely affected by tumor exploitation by producing immunosuppressive and pro-angiogenic factors such as IL-10, arginase 1 (ARG1), TGF-β, or vascular endothelial growth factors (VEGFs), which stimulate tumor cell proliferation, invasion, metastasis, and angiogenesis [41]. Therefore, M2 macrophages are considered a tumor-supporting macrophage phenotype[47].
Tumor-associated macrophages (TAMs)
TAMs are a collective term for macrophages that are prevalent in tumors and can account for up to 50% of some solid tumors [48]. TAMs also share the markers of M1/M2 macrophages [49], however, TAMs rarely exhibit a true M1 or M2 phenotype and are more aptly referred to as M1-like/M2-like TAMs [50]. Under the effects of tumor-secreted colony-stimulating factor 1 (CSF-1, or M-CSF), TAMs polarize to M2-like, allowing immunosuppressive M2-like TAMs to predominate in tumors [47, 51]. High infiltration of M2-like TAMs reduces therapeutic efficacy, shaping tumor-supportive TME, angiogenesis, fibrosis, immunosuppressive cell recruitment, lymphocyte rejection, drug resistance, invasion, and metastasis to enhance tumor progression [52,53,54], which are often associated with poor clinical outcomes [55,56,57].
TAMs are effective target cells in immunotherapy of tumors [12, 58]. This is because macrophages exert opposite anti-tumor or pro-tumor functions through a range of activation pathways and/or different macrophage populations [13, 59]. Different approaches can be taken to eliminate tumor-promoting macrophages and activate or transform them into tumor-suppressing macrophages. Common therapeutic strategies are inhibition of TAMs recruitment [60, 61], reprogramming of TAMs to an M1-like phenotype [62,63,64], and depletion of TAMs [65, 66].
Interaction of OVs, macrophages, and tumors
Macrophage plasticity influences tumor progression and treatment outcome and has a similar effect in oncolytic virotherapy. When OVs are delivered to the body, the body triggers innate immunity in response to the “foreign invasion” of viral infection. Monocytes, macrophages and NK cells will recognize and remove some of the OVs and play a certain inhibitory role. However, in this process, macrophages will also act as carriers of OVs to tumor cells. At the same time macrophages enhance polarization toward a pro-inflammatory phenotype, and this local immune response is also critical for initiating initial anti-tumor immunity [67]. Therefore, we need to further comprehend the complex interactions among OVs, macrophages, and tumors (Fig. 3), to elucidate the mechanisms of macrophages that limit or promote the tumoricidal effects of OVs, and to better utilize the advantages of macrophages to enhance the anti-tumor benefits in future oncolytic virus therapeutic strategies.

Interaction of OVs, macrophages, and tumor cells. After OVs are delivered, some OVs are attacked by activated monocytes/macrophages, causing the viral titer of OVs to decrease. Another portion of OVs can be transported to the tumor site for viral replication, lysing tumor cells and releasing viral progeny, damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and tumor-associated antigens (TAAs). Antigen-presenting cells (APCs) take up and present these antigens, and the resulting activated antigen-specific CD8 + T cells as well as natural killer (NK) cells exert antitumor effects. Secreted IFN-γ and PAMPs repolarize pro-tumorigenic M2-like macrophages into anti-tumorigenic M1-like macrophages, and the anti-tumor/viral effects of the immune system can be further enhanced by secreting IFN-γ and TNF-α
Macrophages limit the antitumor effect of OVs
In general, macrophages show antiviral activity in the setting of oncolytic virotherapy, which is consistent with their defense against pathogens.
Among the routes of administration of OVs, intravenous has more potential than intra-tumoral injection in the treatment of systemic metastatic tumors. However, intravenously administered OVs are often hindered by circulating and tissue immune complexes, neutralizing antibodies, and innate immune cells before reaching the tumor site. Activated macrophages have multiple viral clearance mechanisms, including virus recognition through PRRs, cytokine responses such as IFN, phagocytosis, and activation of other immune cells to reduce viral titers delivered to the tumor site [68, 69].
In a glioma model, phagocytosis by macrophages limits the spread of OVs. Delivery of oncolytic herpes simplex virus (oHSV) after the depletion of macrophages can increase viral titers at tumor sites [70]. IFN and TNF-α signaling is an important mechanism for the antiviral effects of macrophages [71, 72]. In ovarian and breast cancer models it was shown that macrophages can activate the tumor cell JAK/STAT pathway and upregulate the expression of interferon-stimulated genes (ISGs), with tumor cells thereby acquiring an antiviral status that makes them resistant to OVs [73]. In a study of glioblastoma (GBM) treated with oHSV, macrophages, and microglia were found to be the main producers of TNF-α, which inhibits viral replication. Brief administration of TNF-α blockers effectively enhances the killing of tumor cells while reducing inflammation-induced neurotoxicity, enhancing viral replication and survival in GBM intracranial tumors [69]. TAMs and microglia in malignant gliomas largely limit the activity of OVs [74].
Although inflammatory cytokines and phagocytosis produced by macrophages are powerful weapons to kill tumor cells, they also reduce the efficiency of transport of OVs to tumors, so direct delivery of OVs requires a larger viral load to counteract this clearance effect and increases the viral titer of transport to tumor sites.
Macrophages promote the antitumor effect of OVs
However, on the other hand, the interaction between macrophages and OVs could enhance the antitumor effect.
First of all, macrophages can act as carriers of OVs for transport. Macrophages have shown antiviral effects to some extent, but interestingly, increasing studies have evidenced that viruses can utilize monocytes/macrophages as vectors for spreading and replication [75], and macrophages may be an integral part of the therapy of OVs, possibly due to the higher susceptibility of monocytes or naïve macrophages to OVs [76]. Previous research has found that monocytes/macrophages in peripheral blood can act as viral vectors, transporting viable viral particles to tumor sites. Follow-up after intravenous administration of the eutherian virus recovered replicative and oncolytic eutherian virus in blood mononuclear cells even in the presence of neutralizing antibodies (nAbs) to the virus [77]. In another study with oncolytic adenovirus, it was shown that, possibly due to the very low expression of viral antigens, macrophages can act as silent vectors that hide and support viral replication, allowing adenovirus delivery to the tumor site and produce a long-lasting therapeutic effect [78]. More interestingly, recent preclinical studies have found that macrophages are not only capable of uptake and delivery of the tumor oncolytic virus HSV1716 but also support HSV1716 replication within macrophages, which could enhance the effect of viral therapy [79].
Second, OVs can enhance the phagocytic activity of macrophages on tumor cells. As mentioned earlier, TAMs are an important component of macrophages. Activation of TAMs to produce phagocytic activity is a novel mechanism of tumor killing [80], which can be activated by oncolytic virus treatment. CD47 is a membrane-bound protein that is highly expressed on tumor cells and binds to signal regulatory protein α (SIRPα) on macrophages, delivering a “don't eat me” signal that leads to immune evasion by the tumor [81]. After OVs infect cells, PAMPs are exposed to the host immune system, inducing endoplasmic reticulum stress and ICD, leading to the release of DAMPs [82,83,84], which include calreticulin (CRT). CRT, an endoplasmic reticulum-associated molecular chaperone, can also block the CD47 receptor on tumor cells, thereby reducing the “don't eat me” signals generated by macrophages and DCs in response to CD47 binding, and attenuating immune evasion by tumor cells [85]. In addition, after OVs interacted with the B cell receptor (BCR), activated B cells were able to release neutralizing antibodies that mediated NK cell antibody-dependent cytotoxicity (ADCC) and macrophage antibody-dependent cell phagocytosis (ADCP) of virus-infected tumor cells, activating phagocytosis of tumor cells by innate immune cells [86].
Most importantly, OVs can induce polarization of TAMs towards an anti-tumor phenotype. OVs induce activation of NK cells and macrophages through PRRs recognizing PAMPs and DAMPs, secretion of inflammatory cytokines such as IFN-γ, and induced macrophage polarization to M1-like, which results in diminished immunosuppression of TAMs [76, 87]. In an in vitro model of breast cancer, it was found that irrespective of the initial polarization state of macrophages, treatment with oncolytic measles virus (MeV) and mumps virus (MuV) resulted in a significant increase in the M1 macrophage marker, CD80, in human monocyte-derived macrophages (MDMs), while inducing anti-tumor cytokines IL-1β, TNF-α, CXCL9, CXCL10, and IL -6 concentrations were elevated [88]. Preclinical and clinical studies in gastric cancer or glioma have found that treatment with HSV-1 or oncolytic adenovirus rapidly recruited inflammatory cells to the injected lesions, significantly increased the intra-tumoral infiltration of M1-like macrophages and NK cells, with a reduction in the expression of M2-like macrophages, and a significant elevation of the pro-inflammatory cytokines IFN-γ and TNF-α [89, 90]. Although oncolytic adenovirus shifts human macrophages from a more pro-tumor phenotype to a less favorable phenotype, this phenotypic shift is not complete and the M2 trait is not completely lost at the level of gene expression, immunophenotype, and cytokines, which is consistent with the concept that the M1/M2 typing of macrophages is not completely extreme, but rather sequential in phenotype and function [91].
Enhance the synergistic anti-tumor effect of OVs and macrophages
Due to the multifaceted effects generated by macrophages in the treatment of OVs, eliminating the limiting effect of macrophages on OVs, exploiting the effectiveness of macrophages, and obtaining better therapeutic results require intensive research. The current directions are mainly the following: (1), arming OVs to enhance the beneficial effects (pro-inflammatory phenotypic polarization and phagocytosis) or attenuate the adverse effects (antiviral and pro-tumorigenic effects); (2), combining with other drugs to increase the antitumor efficacy; and (3), augmenting the targeting of OVs to tumor cells through effective carrier delivery.
Arming of OVs
OVs can be genetically engineered to arm viruses, and different immunomodulatory genes for arming OVs are being actively tested. Various OVs expressing pro-inflammatory cytokines, chemokines, and other immune checkpoint-associated molecules have been developed to enhance the anti-tumor effects of macrophages (Fig. 4A).

Basic macrophage strategies in oncolytic virotherapy. Currently, there are two major directions of basic strategies for targeting the macrophage to optimize therapeutic response. On the one hand, armed OVs enhance the anti-tumor effect of macrophages. A Repolarization to an antitumor phenotype. Given the pro-tumorigenic role of M2-like tumor-associated macrophages (TAMs), the expression of pro-inflammatory cytokines or chemokines by genetically modified viruses was used to increase macrophage activity and promote the polarization of M2-like macrophages to M1-like macrophages. B Enhancement of phagocytosis by macrophages. The expression of anti-CD47 antibody or SIRPα-Fc fusion protein after viral genetic modification can disrupt “don't eat me” signaling and enhance the killing of tumor cells by macrophages. On the other hand, weakening the clearance of OVs by macrophages contributes to higher viral titers at tumor sites. C Direct macrophage depletion. Since OVs are subject to phagocytosis by macrophages and/or clearance by antiviral cytokines after delivery, brief administration of macrophage depletion agents prior to OVs treatment can cause apoptosis of macrophages, increase the titer of OVs, and change the phenotype of TAMs. D Delivered through the carrier. In addition, the use of tumorophilic carrier cells or liposomes to deliver OVs, is also able to avoid the negative effects of neutralizing antibodies and/or innate immune cells and overcome the challenges of systemic administration of OVs
Arming of OVs to enhance macrophage repolarization
A high M2/M1 ratio in TAMs is strongly associated with tumor progression and poor prognosis. Although OVs can inherently promote polarization of M1-like TAMs and reduce the number of M2-like TAMs, armed OVs can further enhance this polarization.
Talimogene laherparepvec (T-VEC), a GM-CSF-expressing HSV-1, is the first OVs approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with advanced melanoma, with favorable safety and therapeutic outcomes [92]. This is due to the ability of GM-CSF-expressing OVs to attract monocytes and differentiate them into macrophages and DCs, repolarize TAMs from an M2-like phenotype to an M1-like phenotype, and increase the expression of the pro-inflammatory cytokines TNF-α, IL-6, and IL-10 [93, 94].
IL-12 is one of the major regulators of anti-tumor immune responses, promoting the maturation of NK cells, DCs, and T cells, inducing M1-like polarization of macrophages, and increasing IFN-γ levels [95]. Many OVs are currently modified and produce IL-12 [96], and in a GBM model, the use of an oHSV expressing murine IL-12 (G47Δ-mIL12) increased polarization of M1-like TAMs (iNOS+ and pSTAT1+), which may be due to IL-12-induced increases in IFN-γ in the TME [97].
Although IL-12 can effectively induce antitumor immunity, it has certain toxic side effects after systemic administration [95], and IL-21 may be a safer cytokine compared to IL-12. In a pancreatic cancer model study, it was demonstrated that treatment with VVL-21, an oncolytic vaccinia virus (VV) that expresses IL-21, increased the expression of M1-like macrophage marker major histocompatibility complex II (MHC II) and cytokine gene transcripts (IL-6/IL-12 and COX2), and decreased the expression of M2 macrophage marker (CD206) and cytokine gene transcripts (IL-10, TGF-β, and CCL22) expression while also increasing M1 polarization in naïve macrophages [98]. In addition, an IL-36γ-expressing VV (IL-36γ-OVs) was developed. It induces infiltration of lymphocytes and DCs, reduces MDSCs and M2-like TAMs, and has shown significant therapeutic effects in a variety of mouse tumor models [99].
OVs with chemokines are able to effectively recruit immune cells with antitumor effects to migrate to infected tumor sites. Chemokine CC motif ligand 5 (CCL5) promotes immune cell chemotaxis by interacting with chemokine CC motif receptor 1 (CCR1), CCR3, and CCR5 [100]. Infection of tumor cells with CCL5-expressing OVs significantly enhances the migration and activation of NK cells, macrophages, and T cells, and also activates the secretion of CXCL9 by macrophages and DCs aggregated in tumors by binding to tumor cells to activate Fc receptor-mediated ADCC in NK cells and ADCP in macrophages [101, 102], which in turn further promotes the infiltration of circulating T cells into tumor tissues [103].
Both CD40 and OX40 and their ligands CD40L and OX40L belong to the TNF receptor superfamily (TNFRSF). The interaction of CD40 and CD40L activates APCs [104], and the interaction of OX40 and OX40L activates T cells [105], which promotes antitumor effects through activated downstream signaling pathways. A CD40L-expressing oncolytic adenovirus (TMZ-CD40L) is effective in treating pancreatic cancer, a tumor with a high level of M2 macrophages, by increasing the infiltration of M1-like macrophages and T cells into the tumor, repolarizing M2-like macrophages, and controlling tumor progression [106]. Also in a pancreatic cancer model, the use of HSV-1 expressing murine OX40L ((OV-mOX40L) triggered an OX40-OX40L signaling pathway-mediated response that also reprogrammed macrophages and neutrophils to an anti-tumor state, enhanced the anti-tumor response of T cells, and significantly prolonged the survival time of mice [107].
Arming of OVs to enhance phagocytosis
At the same time, it is desired to modify OVs to further block the immunosuppressive effect and enhance phagocytosis of tumors by macrophages (Fig. 4B). An engineered oHSV equipped with a full-length anti-CD47 antibody can be used to disrupt the “don't eat me” signaling generated by the CD47/SIRPα pathway. This oHSV activated phagocytosis and cytotoxicity of tumor cells by macrophages and NK cells, prolonging the survival of glioblastoma and ovarian cancer model mice [108, 109]. Accordingly, investigators designed a VV capable of expressing a chimeric molecule (SIRPα-Fc) consisting of the ectodomain of SIRPα and the Fc structural domain of IgG4. SIRPα-Fc was able to disrupt CD47/SIRPα interactions by blocking CD47 in tumor cells, redirecting macrophages to the tumor site and killing the tumor cells. This VV exerted potent anti-tumor activity in a mouse model of osteosarcoma and can be broadly applied to tumors expressing CD47 [110].
Recently, in a study on cholesterol metabolism, progress has also been made in relation to macrophage phagocytic activity. This study found that TAMs in GBM accumulate cholesterol abnormally, leading to dysfunctional phagocytosis [111]. Apolipoprotein A1 (ApoA1) is a cholesterol reverse transporter protein that allows cholesterol efflux from TAMs, thereby restoring their phagocytosis and antigen-presenting role. Therefore, the investigators developed an ApoA1-expressing oncolytic adenovirus (AdVAPOA1) to intervene in cholesterol metabolism in GBM. AdVAPOA1 activated the TAM-T cell axis and downregulated immune checkpoints after intra-tumor administration, inducing systemic tumor-specific immune memory [111]. This study proposes an immunometabolic treatment approach to armed OVs.
Arming OVs to attenuate the adverse effects of macrophages
Genetically modified OVs not only enhance anti-tumor immunity in macrophages, but also circumvent the detrimental effects of macrophages, including reducing M2-like TAMs and attenuating macrophage-restricted effects on OVs.
Currently, a panel of oncolytic adenoviruses (EnAd) expressing bivalent T-cell engagers (BiTEs) has been designed to target the immunosuppressive effects of M2-like TAMs. The BiTEs recognize CD3ε on T cells and CD206 or folate receptor β (FRβ) on M2-like macrophages. Use of such OVs in patients with malignant ascites activates T cells to selectively kill M2-like macrophages, thereby preserving M1-like macrophages and repolarizing the microenvironment toward a pro-inflammatory state [112].
Human species C adenovirus (HAdv-C5) is bound by immunoglobulin M (IgM) and coagulation factor X (FX) in the blood when delivered intravenously [113, 114], leading to the sequestration of OVs in liver-resident macrophages (Kupffer cells), limiting their tumor targeting and leading to hepatotoxicity [115]. Based on these, the investigators constructed the HAdv-C5 capsid-modified viral variant Ad5-3 M. Ad5-3 M is resistant to IgM- and complement-mediated inactivation, reduces internalization of the viral variant by Kupffer cells, and circumvents the adverse effects of innate immunity to OVs. In mice with disseminated lung tumors, Ad5-3 M prolonged survival and improved safety and efficacy after intravenous administration of OVs [116]. Therefore, the use of genetic modification to change some protein sites in OVs to enhance their resistance is also a worthy direction.
Combination therapy with OVs
In addition to modifying the OVs' own properties, finding the appropriate drugs for combination therapy opens up more possibilities. These strategies include combining immune checkpoint inhibitors to enhance antitumor effects, and combining macrophage depleting agents or immunosuppressive drugs to increase the titer of OVs.
Combination therapy with OVs and immune checkpoint inhibitors (ICIs) is a common combination strategy in clinical trials today (Table 1), due to the ability of OVs to increase the sensitivity of tumor cells to ICIs, which has demonstrated a strong therapeutic effect in a wide range of tumor treatments [117,118,119]. In a GBM model, the use of IL-21-expressing VV (VVDTK-STCDN1L-mIL21) in combination with systemic anti-programmed death receptor 1 (anti-PD1) therapy showed significant induction of M1-like macrophage polarization in the tumor during treatment, along with increased activation of M0 macrophages (MHC II+) in the spleen and DCs in the lymph nodes [120]. Similarly, in other GBM and triple-negative breast cancer models, combination treatment of engineered OVs with ICIs such as anti-cytotoxic T-lymphocyte-associated protein 4 (anti-CTLA-4) antibody, anti-PD-1 antibody and anti-programmed cell death ligand 1 (anti-PD-L1) significantly inhibited tumor growth. The results showed an increase in the proportion of M1-like TAMs, CD4+ and CD8+ cells, and a decrease in the number of immunosuppressive cells such as Tregs. The application of ICIs prevented immune escape from the tumor and overcame the immunosuppressive microenvironment, which is of great significance for the effective eradication of the tumor [97, 121].
OVs combined with macrophage-depleting agents have been reported to remodel TME. In macrophage-dependent tumors, investigators tested the effectiveness of clodronate liposomes and trabectedin in the oHSV treatment of Ewing's sarcoma [122]. Clodronate liposomes can transiently deplete macrophages throughout the body and have demonstrated their therapeutic potential in applications in a variety of tumors [70, 123]. Trabectedin is a chemotherapeutic agent that depletes monocytes/macrophages, including TAMs, by activating caspase-8-dependent apoptosis through the TRAIL receptor [65]. Both drugs were found to enhance antitumor efficacy after macrophage depletion (Fig. 4C). Clodronate liposomes induced antitumor gene expression in TAMs, trabectedin lowered the number of intratumoral MDSCs and M2-like macrophages, and the combination of both drugs with OVs significantly changed the phenotype of TAMs and tended the immune microenvironment to an inflammatory state [122].
Inhibition of macrophage-associated pathways has also shown good efficacy in combination with other immunologic agents. The phosphatidylinositol-3-kinase (PI3K) pathway has an important part in tumor development. PI3K signaling is a key driver of macrophage M2 polarization [124, 125]. PI3Kδ, one of the classes I PI3K isoforms, is hyper-enriched in leukocytes, of which macrophages are included [126]. Some investigators have demonstrated that treatment with PI3Kδ inhibitors prior to intravenous delivery of VV significantly improves VV delivery to tumors and enhances tumor efficacy. This was achieved by interfering with the RhoA/ROCK, AKT, and Rac signaling pathways to inhibit viral attachment to macrophages, independent of viral internalization by macrophages [127]. They combined a PI3Kδ inhibitor (CAL-101), engineered VV, and α-PD1 for the treatment of pancreatic cancer in mice, and the results showed strong synergistic effects, demonstrated the effectiveness of systemic administration, and broke through a major limitation in the treatment of OVs [98]. In addition to this, the use of rapamycin in oncolytic virotherapy has added new possibilities. Rapamycin has immunosuppressive properties and it is able to reduce type I IFN production by inhibiting mammalian target of rapamycin complex 1 (mTORC1) [128], reduce infiltration of CD68+ microglia and CD163+ macrophages in gliomas, and increase viral replication and therapeutic efficacy within tumors [129].
Delivery by carrier
Although suppression of the antiviral immune response of macrophages is beneficial in enhancing the therapeutic effect of OV, such immunosuppression may impair the functional balance of macrophages in vivo and diminish the effect of virus-mediated immune stimulation against cancer. Delivery of OVs using carrier cells with tumorophilic properties can effectively avoid the influence of the immune system and reduce the neutralization and clearance of OVs before they reach the tumor (Fig. 4D). Therefore, this approach may be a more desirable strategy to improve the pharmacokinetics and biological distribution of OVs and has been extensively studied in carrier cells such as mesenchymal stem cells (MSCs), T cells, myeloid cells, and neural stem cells [130].
Moreover, the use of tumor cell tropism to enhance tumor targeting has also been studied accordingly. Membrane-encapsulated oncolytic adenovirus from cancer cells delivered intravenously was able to effectively avoid the antiviral effects of neutralizing antibodies and the innate immune system. This system increases viral replication and enhances the ability of macrophages and DCs to present tumor antigens, and has shown good efficacy in the treatment of different mouse tumor models [131]. When using VV in hosts with pre-existing antibodies to poxviruses, the transient use of a combination of multiple immunosuppressive drugs and cancer cells as carrier cells significantly improves therapeutic efficacy. Although this approach is achieved by increasing the polarization of immunosuppressive M2-like TAMs, such changes are necessary in the long run [132].
Encapsulation of OVs via liposomes (LPs) is also one of the attractive nano-delivery systems. Encapsulation of oncolytic adenovirus (Ad[I/PPT-E1A]) into liposomes coupled to chemokine CC motif ligand 2 (CCL2), which upon intravenous delivery binds to circulating monocytes expressing chemokine CC motif receptor 2 (CCR2), takes advantage of the aggregation of monocytes to hypoxic tumor vessels to deliver encapsulated OVs targeting tumor sites [133]. This system can avoid recognition and delivery to the tumor site by the immune system after intravenous delivery, reducing the number of TAMs located near the blood vessels [134].
Therefore, the use of carriers for adjuvant delivery of OVs is one of the promising strategies. This approach evades the capture of OVs by innate immune cells without affecting the body's immune function, while enhancing the targeting of tumors and reducing the viral delivery load.
In conclusion, macrophages are an important factor affecting the therapeutic effect of OVs, and in the face of this dual effect, how to seek benefits and avoid harm is something we need to consider.
Conclusion and prospect
In conclusion, the wide systemic distribution, plasticity and complex functions of macrophages make them largely influence the outcome and prognosis during oncolytic virotherapy. In the process of tumor development, M1 phenotype macrophages have anti-tumor effects and M2 phenotype macrophages have pro-tumor effects; in oncolytic virotherapy, macrophages can both enhance the effect of viral therapy and limit the spread of OVs to a certain extent. These different regulatory effects may be related to tumor heterogeneity, the type of OVs, and host responsiveness [8]. The specific applications of these different types of OVs in different tumors need to be emphasized. At the same time, facing this dual role, how to seek benefits and avoid harm is a key concern that we need to focus on.
For the aspect of enhancing the beneficial effects of macrophages, gene-regulated OVs demonstrated a strong “re-education” capacity. Various cytokines, chemokines, and immunomodulatory antibodies remodel TAMs and enhance the anti-tumor effects of macrophages. This is one of the modalities with infinite exploits and possibilities that need to be constantly developed and added to.
For the aspect of attenuating the harmful effects of macrophages, we need interventions to avoid unwanted immune responses; after all, intravenous delivery of OVs is a much more promising and ubiquitous mode of administration than local delivery. Direct strategies include the use of immunosuppressive or macrophage-depleting agents for short periods of time to inhibit macrophage capture of OVs. Indirect strategies include the use of genetic modification of viruses to avoid antibody-mediated macrophage clearance and the use of carrier cells or other nanomaterial encapsulation to overcome barriers to intravenous delivery.
We also need to further examine the effect of antigen expression of various viruses on the activation of host immunity, and modification of OVs with extremely low antigen expression without loss of oncolytic biological activity is a major challenge for intravenous delivery. Therefore, we had to elucidate more deeply the interactions among OVs, macrophages, and tumor cells and their mechanisms of action, and utilize the powerful regulatory function of macrophages to synergistically enhance the application of OVs. In future clinical trials of oncolytic virotherapy, it is particularly important to optimize intravenous delivery, avoid unnecessary immune effects, reduce the delivery load of viruses, improve the oncolytic effect of OVs, and enhance the combined therapeutic effect to ultimately achieve a durable and effective treatment outcome.
Data availability
Not applicable.
Abbreviations
- OVs:
-
Oncolytic viruses
- TME:
-
Tumor microenvironment
- TAMs:
-
Tumor-associated macrophages
- IFN:
-
Interferon
- ICD:
-
Immunogenic cell death
- TNF-α:
-
Tumor necrosis factor-α
- IL:
-
Interleukin
- DCs:
-
Dendritic cells
- PAMPs:
-
Pathogen-associated molecular patterns
- DAMPs:
-
Damage-associated molecular patterns
- TAAs:
-
Tumor-associated antigens
- PRRs:
-
Pattern recognition receptors
- TLRs:
-
Toll-like receptors
- APCs:
-
Antigen-presenting cells
- Tregs:
-
Regulatory T cells
- TRMs:
-
Tissue-resident macrophages
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- CXCL:
-
Chemokines CXC motif ligand
- M-CSF:
-
Macrophage colony-stimulating factor
- TGF:
-
Transforming growth factor
- CSF -1:
-
Colony-stimulating factor 1
- HSV:
-
Herpes simplex virus
- GBM:
-
Glioblastoma
- SIRPα:
-
Signal regulatory protein α
- MDMs:
-
Monocyte-derived macrophages
- T-VEC:
-
Talimogene laherparepvec
- VV:
-
Vaccinia virus
- CCL:
-
Chemokine CC motif ligand
- CCR:
-
Chemokine CC motif receptor
- HAdv-C5:
-
Human species C adenovirus
- ICIs:
-
Immune checkpoint inhibitors
References
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70.
O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67.
Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11(4):933–59.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–86.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22.
Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218.
Jafari M, Kadkhodazadeh M, Shapourabadi MB, et al. Immunovirotherapy: the role of antibody based therapeutics combination with oncolytic viruses. Front Immunol. 2022;13:1012806.
Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol. 2018;39(3):209–21.
Wan PK, Ryan AJ, Seymour LW. Beyond cancer cells: Targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Mol Ther. 2021;29(5):1668–82.
Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol. 2017;7:106.
Sheu KM, Hoffmann A. Functional hallmarks of healthy macrophage responses: their regulatory basis and disease relevance. Annu Rev Immunol. 2022;40:295–321.
Liu L, Li H, Wang J, et al. Leveraging macrophages for cancer theranostics. Adv Drug Deliv Rev. 2022;183: 114136.
Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021;81(5):1201–8.
Jakeman PG, Hills TE, Fisher KD, Seymour LW. Macrophages and their interactions with oncolytic viruses. Curr Opin Pharmacol. 2015;24:23–9.
Davola ME, Mossman KL. Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? Oncoimmunology. 2019;8(6): e1581528.
Li M, Li G, Kiyokawa J, et al. Characterization and oncolytic virus targeting of FAP-expressing tumor-associated pericytes in glioblastoma. Acta Neuropathol Commun. 2020;8(1):221.
Rahman MM, McFadden G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers. 2021;13(21):5452.
Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019;18(9):689–706.
Wang JY, Chen H, Dai SZ, et al. Immunotherapy combining tumor and endothelium cell lysis with immune enforcement by recombinant MIP-3α Newcastle disease virus in a vessel-targeting liposome enhances antitumor immunity. J Immunother Cancer. 2022;10(3): e003950.
Tian Y, Xie D, Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct Target Ther. 2022;7(1):117.
Peters CWD, Nigim F. Immunomodulatory arming factors-the current paradigm for oncolytic vectors relies on immune stimulating molecules. Int J Mol Sci. 2021;22(16):9051.
Ilkow CS, Swift SL, Bell JC, Diallo JS. From scourge to cure: tumour-selective viral pathogenesis as a new strategy against cancer. PLoS Pathog. 2014;10(1): e1003836.
Hindupur SV, Schmid SC, Koch JA, et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int J Mol Sci. 2020;21(3):1106.
Ma R, Li Z, Chiocca EA, Caligiuri MA, Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. 2023;9(2):122–39.
Ma J, Ramachandran M, Jin C, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020;11(1):48.
Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41(3):421–33.
Zhu Z, McGray AJR, Jiang W, Lu B, Kalinski P, Guo ZS. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol Cancer. 2022;21(1):196.
Garmaroudi GA, Karimi F, Naeini LG, Kokabian P, Givtaj N. Therapeutic efficacy of oncolytic viruses in fighting cancer: recent advances and perspective. Oxid Med Cell Longev. 2022;2022:3142306.
Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27(8):1482–92.
Friedman GK, Johnston JM, Bag AK, et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med. 2021;384(17):1613–22.
Wang L, Chard Dunmall LS, Cheng Z, Wang Y. Remodeling the tumor microenvironment by oncolytic viruses: beyond oncolysis of tumor cells for cancer treatment. J Immunother Cancer. 2022;10(5): e004167.
Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. 2019;19(5):291–304.
Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol. 2017;79:593–617.
Wang S, Yang Y, Ma P, et al. Landscape and perspectives of macrophage -targeted cancer therapy in clinical trials. Mol Ther Oncolytics. 2022;24:799–813.
Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–47.
Nobs SP, Kopf M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol. 2021;42(6):495–507.
Liu Z, Gu Y, Chakarov S, et al. Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell. 2019;178(6):1509-1525.e19.
Park MD, Silvin A, Ginhoux F, Merad M. Macrophages in health and disease. Cell. 2022;185(23):4259–79.
Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. 2022;23(8):1148–56.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–82.
Orekhov A, Orekhova V, Nikiforov N, et al. Monocyte differentiation and macrophage polarization. Vessel Plus. 2019;3:10.
van Dalen FJ, van Stevendaal MHME, Fennemann FL, Verdoes M, Ilina O. Molecular repolarisation of tumour-associated macrophages. Molecules. 2018;24(1):9.
Mills CD, Lenz LL, Harris RA. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res. 2016;76(3):513–6.
Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs functional differentiation. Front Immunol. 2014;5:514.
Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. 2018;19(6):1801.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37.
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor Microenvironment. Cell Metab. 2019;30(1):36–50.
Müller S, Kohanbash G, Liu SJ, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234.
Pittet MJ, Michielin O, Migliorini D. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol. 2022;19(6):402–21.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55.
Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6(1):75.
Xie Y, Yang H, Yang C, et al. Role and mechanisms of tumor-associated macrophages in hematological malignancies. Front Oncol. 2022;12: 933666.
Zhou Z, Peng Y, Wu X, et al. CCL18 secreted from M2 macrophages promotes migration and invasion via the PI3K/Akt pathway in gallbladder cancer. Cell Oncol. 2019;42(1):81–92.
Cao Y, Qiao B, Chen Q, et al. Tumor microenvironment remodeling via targeted depletion of M2-like tumor-associated macrophages for cancer immunotherapy. Acta Biomater. 2023;160:239–51.
Zheng X, Weigert A, Reu S, et al. Spatial density and distribution of tumor-associated macrophages predict survival in non-small cell lung carcinoma. Cancer Res. 2020;80(20):4414–25.
Cortese N, Carriero R, Laghi L, Mantovani A, Marchesi F. Prognostic significance of tumor-associated macrophages: past, present and future. Semin Immunol. 2020;48: 101408.
Tzetzo SL, Abrams SI. Redirecting macrophage function to sustain their “defender” antitumor activity. Cancer Cell. 2021;39(6):734–7.
Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15(12):731–44.
Wang H, Shao Q, Wang J, et al. Decreased CXCR2 expression on circulating monocytes of colorectal cancer impairs recruitment and induces Re-education of tumor-associated macrophages. Cancer Lett. 2022;529:112–25.
Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112–23.
Kaneda MM, Messer KS, Ralainirina N, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–42.
Rodell CB, Arlauckas SP, Cuccarese MF, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578–88.
Zeng Q, Jewell CM. Directing toll-like receptor signaling in macrophages to enhance tumor immunotherapy. Curr Opin Biotechnol. 2019;60:138–45.
Germano G, Frapolli R, Belgiovine C, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62.
Cotechini T, Atallah A, Grossman A. Tissue-resident and recruited macrophages in primary tumor and metastatic microenvironments: potential targets in cancer therapy. Cells. 2021;10(4):960.
Melcher A, Harrington K, Vile R. Oncolytic virotherapy as immunotherapy. Science. 2021;374(6573):1325–6.
Vitiello GAF, Ferreira WAS, Cordeiro de Lima VC, Medina TDS. Antiviral responses in cancer: boosting antitumor immunity through activation of interferon pathway in the tumor microenvironment. Front Immunol. 2021;12:782852.
Meisen WH, Wohleb ES, Jaime-Ramirez AC, et al. The impact of macrophage- and microglia-secreted TNFα on oncolytic HSV-1 therapy in the glioblastoma tumor microenvironment. Clin Cancer Res. 2015;21(14):3274–85.
Fulci G, Dmitrieva N, Gianni D, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67(19):9398–406.
Kong X, Lu X, Wang S, et al. Type I interferon/STAT1 signaling regulates UBE2M-mediated antiviral innate immunity in a negative feedback manner. Cell Rep. 2023;42(1): 112002.
Josephs SF, Ichim TE, Prince SM, et al. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J Transl Med. 2018;16(1):242.
Liu YP, Suksanpaisan L, Steele MB, Russell SJ, Peng KW. Induction of antiviral genes by the tumor microenvironment confers resistance to virotherapy. Sci Rep. 2013;3:2375.
Zhang Q, Liu F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis. 2020;11(6):485.
Nikitina E, Larionova I, Choinzonov E, Kzhyshkowska J. Monocytes and macrophages as viral targets and reservoirs. Int J Mol Sci. 2018;19(9):2821.
Polzin M, McCanless J, Owen S, et al. Oncolytic vesicular stomatitis viruses selectively target M2 macrophages. Virus Res. 2020;284: 197991.
Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4(138):138ra77.
Muthana M, Giannoudis A, Scott SD, et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011;71(5):1805–15.
Kwan A, Winder N, Atkinson E, et al. Macrophages mediate the antitumor effects of the oncolytic Virus HSV1716 in mammary tumors. Mol Cancer Ther. 2021;20(3):589–601.
Lecoultre M, Dutoit V, Walker PR. Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: a review. J Immunother Cancer. 2020;8(2): e001408.
Weiskopf K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur J Cancer. 2017;76:100–9.
Garg AD, Agostinis P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol Rev. 2017;280(1):126–48.
Thomas S, Kuncheria L, Roulstone V, et al. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer. 2019;7(1):214.
Bommareddy PK, Zloza A, Rabkin SD, Kaufman HL. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology. 2019;8(7):1591875.
Fucikova J, Spisek R, Kroemer G, Galluzzi L. Calreticulin and cancer. Cell Res. 2021;31(1):5–16.
Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13(1):84.
Burke S, Shergold A, Elder MJ, et al. Oncolytic Newcastle disease virus activation of the innate immune response and priming of antitumor adaptive responses in vitro. Cancer Immunol Immunother. 2020;69(6):1015–27.
Tan DQ, Zhang L, Ohba K, Ye M, Ichiyama K, Yamamoto N. Macrophage response to oncolytic paramyxoviruses potentiates virus-mediated tumor cell killing. Eur J Immunol. 2016;46(4):919–28.
Sugawara K, Iwai M, Yajima S, et al. Efficacy of a third-generation oncolytic herpes virus G47Δ in advanced stage models of human gastric cancer. Mol Ther Oncolytics. 2020;17:205–15.
van Putten EHP, Kleijn A, van Beusechem VW, et al. Convection enhanced delivery of the oncolytic adenovirus delta24-RGD in patients with recurrent GBM: a phase I clinical trial including correlative Studies. Clin Cancer Res. 2022;28(8):1572–85.
van den Bossche WBL, Kleijn A, Teunissen CE, et al. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol. 2018;20(11):1494–504.
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
Chon HJ, Lee WS, Yang H, et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res. 2019;25(5):1612–23.
Hsieh CC, Hsieh MJ, Wang YH, Liao ZX. Macrophage distribution affected by virus-encoded granulocyte macrophage colony stimulating factor combined with lactate oxidase. ACS Omega. 2022;7(27):24020–6.
Tugues S, Burkhard SH, Ohs I, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22(2):237–46.
Nguyen HM, Guz-Montgomery K, Saha D. Oncolytic virus encoding a master pro-inflammatory cytokine interleukin 12 in cancer immunotherapy. Cells. 2020;9(2):400.
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253-267.e5.
Marelli G, Chard Dunmall LS, Yuan M, et al. A systemically deliverable Vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J Immunother Cancer. 2021;9(1): e001624.
Yang M, Giehl E, Feng C, et al. IL-36γ-armed oncolytic virus exerts superior efficacy through induction of potent adaptive antitumor immunity. Cancer Immunol Immunother. 2021;70(9):2467–81.
Pol JG, Workenhe ST, Konda P, Gujar S, Kroemer G. Cytokines in oncolytic virotherapy. Cytokine Growth Factor Rev. 2020;56:4–27.
Tian L, Xu B, Chen Y, et al. Specific targeting of glioblastoma with an oncolytic virus expressing a cetuximab-CCL5 fusion protein via innate and adaptive immunity. Nat Cancer. 2022;3(11):1318–35.
Sanchez Gil J, Rabkin SD. An armed oncolytic virus for GBM destruction. Nat Cancer. 2022;3(11):1274–6.
Ang L, Li J, Dong H, et al. Chimeric oncolytic adenovirus armed chemokine rantes for treatment of breast cancer. Bioengineering. 2022;9(8):342.
Yan C, Richmond A. Hiding in the dark: pan-cancer characterization of expression and clinical relevance of CD40 to immune checkpoint blockade therapy. Mol Cancer. 2021;20(1):146.
Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B. 2020;10(3):414–33.
Eriksson E, Moreno R, Milenova I, et al. Activation of myeloid and endothelial cells by CD40L gene therapy supports T-cell expansion and migration into the tumor microenvironment. Gene Ther. 2017;24(2):92–103.
Liu S, Li F, Ma Q, et al. OX40L-armed oncolytic virus boosts T-cell response and remodels tumor microenvironment for pancreatic cancer treatment. Theranostics. 2023;13(12):4016–29.
Xu B, Tian L, Chen J, et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat Commun. 2021;12(1):5908.
Tian L, Xu B, Teng KY, et al. Targeting Fc receptor-mediated effects and the “don’t eat me” signal with an oncolytic virus expressing an Anti-CD47 antibody to treat metastatic ovarian cancer. Clin Cancer Res. 2022;28(1):201–14.
Cao F, Nguyen P, Hong B, et al. Engineering oncolytic vaccinia virus to redirect macrophages to tumor cells. Adv Cell Gene Ther. 2021;4(2): e99.
Wang S, Yan W, Kong L, et al. Oncolytic viruses engineered to enforce cholesterol efflux restore tumor-associated macrophage phagocytosis and anti-tumor immunity in glioblastoma. Nat Commun. 2023;14(1):4367.
Scott EM, Jacobus EJ, Lyons B, et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J Immunother Cancer. 2019;7(1):320.
Doronin K, Flatt JW, Di Paolo NC, et al. Coagulation factor X activates innate immunity to human species C adenovirus. Science. 2012;338(6108):795–8.
Maloney BE, Perera KD, Saunders DRD, Shadipeni N, Fleming SD. Interactions of viruses and the humoral innate immune response. Clin Immunol. 2020;212: 108351.
Khare R, Hillestad ML, Xu Z, Byrnes AP, Barry MA. Circulating antibodies and macrophages as modulators of adenovirus pharmacology. J Virol. 2013;87(7):3678–86.
Atasheva S, Emerson CC, Yao J, Young C, Stewart PL, Shayakhmetov DM. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci Transl Med. 2020;12(571):eabc6659.
Chiu M, Armstrong EJL, Jennings V, et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Expert Opin Biol Ther. 2020;20(6):635–52.
Bourgeois-Daigneault MC, Roy DG, Aitken AS, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10(422):eaao1641.
Lee YS, Lee WS, Kim CW, et al. Oncolytic vaccinia virus reinvigorates peritoneal immunity and cooperates with immune checkpoint inhibitor to suppress peritoneal carcinomatosis in colon cancer. J Immunother Cancer. 2020;8(2): e000857.
Sun Y, Zhang Z, Zhang C, et al. An effective therapeutic regime for treatment of glioma using oncolytic vaccinia virus expressing IL-21 in combination with immune checkpoint inhibition. Mol Ther Oncolytics. 2022;26:105–19.
Zhang H, Xie W, Zhang Y, et al. Oncolytic adenoviruses synergistically enhance anti-PD-L1 and anti-CTLA-4 immunotherapy by modulating the tumour microenvironment in a 4T1 orthotopic mouse model. Cancer Gene Ther. 2022;29(5):456–65.
Denton NL, Chen CY, Hutzen B, et al. Myelolytic treatments enhance oncolytic herpes virotherapy in models of ewing sarcoma by modulating the immune microenvironment. Mol Ther Oncolytics. 2018;11:62–74.
Opperman KS, Vandyke K, Clark KC, et al. Clodronate-liposome mediated macrophage depletion abrogates multiple myeloma tumor establishment in vivo. Neoplasia. 2019;21(8):777–87.
Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26.
Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198(3):1006–14.
Vanhaesebroeck B, Perry MWD, Brown JR, André F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov. 2021;20(10):741–69.
Ferguson MS, Chard Dunmall LS, Gangeswaran R, et al. Transient inhibition of PI3Kδ enhances the therapeutic effect of intravenous delivery of oncolytic vaccinia virus. Mol Ther. 2020;28(5):1263–75.
Alain T, Lun X, Martineau Y, et al. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci U S A. 2010;107(4):1576–81.
Lun X, Alain T, Zemp FJ, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70(2):598–608.
Reale A, Calistri A, Altomonte J. Giving oncolytic viruses a free ride: carrier cells for oncolytic virotherapy. Pharmaceutics. 2021;13(12):2192.
Fusciello M, Fontana F, Tähtinen S, et al. Artificially cloaked viral nanovaccine for cancer immunotherapy. Nat Commun. 2019;10(1):5747.
Guo ZS, Parimi V, O’Malley ME, et al. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther. 2010;17(12):1465–75.
Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. 2018;15(4):335–45.
Iscaro A, Jones C, Forbes N, et al. Targeting circulating monocytes with CCL2-loaded liposomes armed with an oncolytic adenovirus. Nanomedicine. 2022;40: 102506.
Acknowledgements
Not applicable.
Funding
This work was supported by the Scientific and Technological Innovation Major Base of Guangxi (No.2022-36-Z05); Guangxi Natural Science Foundation for Distinguished Young Scholars (No.GXNSFFA035026); The National Natural Science Foundation of China (No.82104654).
Author information
Authors and Affiliations
Contributions
ZS, XYL, and GXF designed and wrote the original manuscript. JTN and QQL prepared the figures and table, FQL, ZKZ, and LPZ participated in revising and reviewing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have declared no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, Z., Liu, X., Fan, G. et al. Improving the therapeutic efficacy of oncolytic viruses for cancer: targeting macrophages. J Transl Med 21, 842 (2023). https://doi.org/10.1186/s12967-023-04709-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04709-z