
Abstract
The immune system in humans is a defense department against both exogenous and endogenous hazards, where CD8+ T cells play a crucial role in opposing pathological threats. Various immunotherapies based on CD8+ T cells have emerged in recent decades, showing their promising results in treating intractable diseases. However, in the fight against the constantly changing and evolving cancers, the formation and function of CD8+ T cells can be challenged by tumors that might train a group of accomplices to resist the T cell killing. As cancer therapy stepped into the era of immunotherapy, understanding the physiological role of CD8+ T cells, studying the machinery of tumor immune escape, and thereby formulating different therapeutic strategies become the imperative missions for clinical and translational researchers to fulfill. After brief basics of CD8+ T cell-based biology is covered, this review delineates the mechanisms of tumor immune escape and discusses different cancer immunotherapy regimens with their own advantages and setbacks, embracing challenges and perspectives in near future.
Similar content being viewed by others

Introduction
The basics of cancer immunotherapy is to initiate and optimize the key procedures in the innate or acquired immune system, including but not limited to, surveillance, identification, and elimination of tumors [1]. The ultimate executors of acquired cellular immunity, i.e., CD8+ T cells, are exceptionally destructive when encountering tumors [2]. The advent of novel cancer treatments, such as immune checkpoint blockers, neoantigen vaccines, chimeric antigen receptor T (CAR-T) cell therapy and T-cell receptor T (TCR-T) cell therapy, points to that CD8+ T cell-based immunotherapy has gained dramatic breakthroughs. Recently, autologous cell therapy using tumor-infiltrating lymphocytes (TILs) has been approved by the United States (US) Food and Drug Administration (FDA) to treat patients with unresectable or metastatic melanoma, further bolstering the already rapid development of novel CD8+ T cell-involved therapy [3].
Under normal circumstances, the immune system monitors the aberrant activities in the body and identifies and removes the suspicious components (e.g., tumor cells), thereby preventing tumor initiation. However, in patients suffering from cancers, the fulfillment of immune surveillance can go awry. A variety of immune escape strategies might be adopted by tumors to counteract the host’s monitoring, thus leading to uncontrolled tumor growth [4, 5]. Under these circumstances, the effective functions of CD8+ T cells can be remarkably disturbed. Therefore, exploration of constantly changing immune escape mechanisms and development of the emerging CD8+ T cell-based immunotherapy strategies make the driving seats behind the wheel of cancer research (1).
Nevertheless, albeit immunotherapy demonstrated tremendous success in oncological treatment, a relatively small fraction of patients with cancers, especially solid tumors, responded to a diversity of immunotherapies [6, 7]. Concurrently, incidents of adverse events and cancer recurrence following immunotherapy still exist [8,9,10]. Therefore, in this review, we summarize the recent advances on CD8+ T cell-based cancer therapy, aiming to introduce the basics of CD8+ T cells through their generation, activation, function, and destination, together with new research findings, and to delineate the machinery regarding the immune escape of tumors by deceiving CD8+ T cells and immunotherapy regimens based on activation CD8+ T cells, all mingled with challenges and progresses.
The life cycle of CD8+ T cells
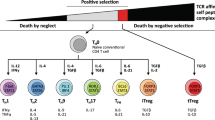
Red bone marrow produces the precursor T cells, which are recruited to the thymus under the action of chemokines [11]. In the thymus, mature CD8+ or CD4+ T cells are transported to the peripheral blood after positive and negative selection with assistance of antigen-presenting cells (APCs), thymic epithelial cells and thymus factors (Fig. 1A) [12]. The complete activation of antigen-specific CD8+ T cells requires the synergy combining three types of signals, including the pioneer signals initiated by TCR binding to peptide-bound major histocompatibility complex (MHC), the ligand-mediated costimulatory signal, and the supplementary signals produced by cytokines [13, 14]. Eventually, the above signals initiate a cascade of effector proteins that accelerate T cell activation (Fig. 1B).

The life cycle of CD8+ T cells. (A) Birth of mature CD8+ T cells. Red bone marrow produces pre-T cells, which are recruited to the thymus under the action of chemokines. Subsequently, T cells undergo positive and negative selection to become mature CD4+T or CD8+T cells, assisted by antigen-presenting cells (APCs), thymic epithelial cells and thymic factors. (B) Activation of CD8+ T cells. The activation of CD8+ T cells requires the assistance of three signals, namely the precursor signal triggered by the binding of TCR and MHC-I, the ligand mediated co-stimulatory signal and the supplementary signal produced by cytokines. ITAM, immunoreceptor tyrosine-based activation motif; LCK, lymphocyte-specific protein tyrosine kinase. (C) Anti-tumor effect of CD8+ T cells. CD8+ T cells can form immune synapses with target cells after activation. Subsequently, CD8+ T cells release granzymes, perforin, and cytokines to destroy tumor cells. Meanwhile, CD8+ T cells express the death receptor Fas-L and induce apoptosis in Fas expressing tumor cells. In addition, activated CD8+ T cells may kill target cells through the delivery of EVs. Tumor antigens, once released, are presented by DCs and T cells are activated to infiltrate tumors, recognizing and dismantling tumor cells. LFA-1, lymphocyte function-associated antigen 1; ICAM, intercellular cell adhesion molecule. (D) The fate of CD8+ T cells. CD8+ T cells expressing death ligand receptors can induce activated cell death of their own or adjacent CD8+ T lymphocytes. Meanwhile, apoptosis related gene 2 can accelerate the CD8+ T cell death by cleaving the differentiation protein of myeloid leukemia cells. The expression of pro-apoptotic proteins BAX and BAK enhances mitochondrial membrane permeability, leading to the activation of cytochrome c and its binding to apoptotic protease activating factor-1, further leading to caspase 8/-3 dependent cell death
Antigen-specific CD8+ T cells are activated after recognizing tumor-associated antigens presented by MHC class I (MHC I) molecules, and an immune synapse is formed between the target tumor cells and CD8+ T cells of high functional and structural avidity that preferentially reside in tumors [15]. A lately study also revealed that neonatal CD8+ T cells could undergo a bystander activation in response to innate cytokines without cognate TCR stimulation [16]. But whether this TCR-independent activation take place in the context of tumor killing remains unknown. Consequently, CD8+ T cells release granzymes, perforins and cytokines through the immune synapse in a high density to destroy tumor cells, and on many occasions this cytotoxicity could be delivered via T cell-derived extracellular vesicles (EVs) [17, 18]. In parallel, CD8+ T cells can express the death receptor Fas-L and induce apoptosis in Fas-expressing tumor cells [19], or kill target cells through other destructive pathways, such as pyroptosis [20, 21] or ferroptosis [22] (Fig. 1C).
As activated CD8+ T cells cannot proliferate or survive indefinitely, their fate is rigorously manipulated by several immune balance mechanisms. Those cells expressing both death receptors and ligands can induce activated cell death of self or adjacent CD8+ T cells [23]. At the same time, CD8+ T cells can maintain their own homeostasis in a death receptor-independent way. For example, apoptosis-related gene 2 can accelerate the death of CD8+ T cells by lysing myeloid leukemia cell differentiation proteins [24]. Pro-apoptotic proteins BAX and BAK are expressed, and the permeability of mitochondrial membrane is enhanced, resulting in the activation of cytochrome c and its binding to apoptotic peptidase activator 1 for further caspase-8/-3-dependent cell death [25]. Additionally, CD8+ T cells can be eliminated through immune checkpoint inhibitory signaling [26], transforming growth factor b (TGF-b) signaling [27] or/and autophagy [28] (Fig. 1D).
Cancer immune escape: in a close relation to CD8+ T cells
Cancer cell
Lack of immunogenicity
Activation of specific CD8+ T cells requires the presence of recognizable tumor antigens, while the lack of immunogenicity for certain antigens may be the reason for the failure of T cell immune response [29]. Cancer cells can downregulate or neutralize self-antigens to lower immunogenicity, so deceiving the immune system [30]. In the early stage of tumor progression, most MHC I molecules on the surface of tumor cells act as a medium for CD8+ T cells to recognize, so to kill them [31]. Unfortunately, a variety of tumor cells downregulate human leukocyte antigen (HLA) genes to reduce MHC I molecules so that the decrease of tumor antigen presentation tricks the CD8+ T cells into skipping their killing [32]. As a result, CD8+ T cells eliminate most of the MHC I-positive tumor cells, while MHC I-negative tumor cells survive and grow [33]. Additionally, genes related to antigen processing such as ER-resident aminopeptidase are frequently downregulated in tumor microenvironment (TME) [34].
Many types of tumor cells can alter their surface glycosylation to evade the immune response of T cells. Those can express sialic acid-carrying glycans to cover the cell membrane, so preventing the maturation of dendritic cells (DCs) in the TME and impairing their antigen-presenting functions [35]. The sialic acid blockers can thus reduce the sialylation, restore the tumor immunogenicity, and promote the aggregation and cytotoxicity of CD8+ T cells to clear tumors [36]. In addition, CD39 is a rate-limiting enzyme in the process of ATP conversion to adenosine, as high expression of CD39 on the tumors surface represses the maintenance of extracellular ATP inflammatory signals, leading to the formation of non-inflammatory tumors and loss of immune response [37].
Secretion of immunosuppressive factors and enzymes
Tumor cells can upregulate the expression of multiple immunosuppressive substances to help their evasion [38]. For example, together with the ATF1 transcription factor, SMAD protein activated by TGF-β can bind and inhibit the promoter genes of the granzyme B and interferon γ (IFN-γ), thereby restraining the cytotoxic function of CD8+ T cells [39]. Concomitantly, TGF-β can diminish expression of chemokine receptor CXC motif 3 (CXCR3) that is primarily expressed on activated CD8+ T cells, by increasing SMAD protein binding to CXCR3 promoter, so restricting their trafficking into tumors [40]. Similarly, the secretion of IL-37b in melanoma cells downregulates the co-stimulatory molecules on DCs, then imposing severe damage on CD8+ T cell activation [41]. However, an atypical example can be IL-10, an anti-inflammatory cytokine, which has the dual role of inhibiting [42] and promoting tumors [43, 44]. IL-10 can suppress myeloid and chronic inflammatory T cell responses and expand tumor specific CD8+ T cells. On the contrary, IL-10 also induces IFN-γ and cytotoxic mediators in antigen-activated T cells [45]. Thus, IL-10 plays a crucial role in the shift from activation to exhaustion in T cells.
Indoleamine 2, 3-dioxygenase 1 (IDO1) is highly expressed on many cancers, being a pivotal enzyme in the pathway of tryptophan metabolism [46]. It has been well established that tryptophan is essential for T cell proliferation and activation [47]. Tryptophan metabolites own effective T-cell-inhibiting capacities and can enhance the differentiation and immunosuppression ability of regulatory T cells (Tregs), thus promoting the progression of several cancers, including colon cancer and pancreatic adenocarcinoma [48, 49]. A study further demonstrated that IDO1 could inhibit the response of CD8+ T cells and promote the tumor growth in subcutaneous colon cancer models, highlighting the role of IDO1 in tumor immune escape [50].
Expression of immunosuppressive coreceptors
Under the assistance of co-stimulation signals, CD8+ T cells can be further activated to accurately regulate the autoimmunity. These co-stimulatory signals deliver suppressing or promoting messages to T cells so that the presence of inhibitory receptors brings the negative regulation of T cell response to avoid damaging autoimmunity [51]. However, untamable tumor cells can frequently raise the expression of regulatory proteins on the cell surface, including cytotoxic T lymphocyte antigen 4 (CTLA-4), Fas-L, programmed cell death-ligand 1 (PD-L1), lymphocyte activation gene-3 (LAG-3) and T cell immunoglobulin and ITIM domain (TIGIT), so triggering T cell apoptosis [52]. For instance, gastric adenocarcinomas infiltrated by high density CD8+ T cells can express high levels of PD-L1 to avoid immune surveillance [53].
Understanding towards several tumor-related signaling pathways, such as Wnt/β-catenin, MAPK, EGFR, and STAT, has uncovered partial mechanisms involved in cancer immune escape [54]. Among them, Wnt/β-catenin signaling pathway has been proven a major route of tumor immunosuppression in colorectal cancer, breast cancer and melanoma [55]. β-catenin can promote the expression of transcription suppressor ATF3 to downregulate CCL4, so to impede the recruitment of DCs and CD8+ T cells into tumors [56]. In parallel, β-catenin promotes Tregs differentiation by stimulating tumor production of IDO1 and synergistically suppressing the immune response [57].
Release of EVs
Tumors can evade immune killing by secreting small EVs (traditionally known as exosomes), containing soluble factors, enzymes, immunoregulatory receptor ligands and RNAs, to interfere with the immune system [58]. Exosomes containing TGF-β released by tumor cells under hypoxic conditions promote the activation of Tregs [59]. Tregs co-cultured with exosomes derived from tumor cells also showed high expressions of Fas-L and CTLA-4, ably inhibiting the function of CD8+ T cells [60].
The arena of tumor exosomes is not limited to TME. For instance, exosomes secreted by melanoma cells are abundant in PD-L1, which can confront CD8+ T cells in the circulatory system and undermine their function. In addition, IFN-γ upregulates PD-L1 in exosomes [60]. Exosomes that originate from melanoma contain a variety of chemokines (CXCL1, CXCL10, CCL2, CCL5) to recruit immunosuppressive cells such as neutrophils [61]. After mRNAs in tumor-derived exosomes are transferred to T cells, the mRNA transcript is finally translated into immunosuppressive proteins, indicating that tumor-derived exosomes can induce immune tolerance through reprogramming [62]. Alternatively, CD39+ Tregs are continuously stimulated by CD73 in tumor-derived exosomes, causing adenosine upregulation [63, 64]. Thus, adenosine interacts with adenosine A2A receptor to rise cAMP level, therefore triggering CD8+ T cell anergy [65].
Tumor suppressive environment
Immunosuppressive cell infiltration
Tregs
Tregs are the primary factors in the formation of immunosuppressive environments in tumors [66]. Myeloid-derived inhibitory cells and tumor cells upregulate the expression of CCL5, CCL22 and CCL28 to attract Tregs expressing CCR5, CCR10 or CCR4 to migrate toward tumors [67]. Tregs can express inhibitory receptors such as CTLA-4, T cell immunoglobulin and mucin-domain containing-3 (TIM-3), PD-1, GITR, LAG-3, BTLA and NRP-1, and secrete active substances such as IL-10, TGF-β, IL-2, IL-35, IDO1 and adenosine, respectively, through various mechanisms to inhibit the differentiation, activation and function of CD8+ T cells [68]. Tregs, an enhancer of immune suppression, inhibit the secretion of anti-inflammatory factors [69] (Fig. 2).

Intratumoral immune cells form a complex network that promotes tumor immune escape. TME hosts infiltrating immune cells, including MDSCs, DCs, mast cells, macrophages, neutrophils, lymphocytes, and natural killer cells with aberrant functions, which may work synergistically in a group to accomplice with tumors to evade immune surveillance and destruction, thereby promoting tumor growth. Reproduced with permission [156]
Tolerogenic DCs
Dendritic cells are professionals who present antigens to CD8+ T cells through MHC I molecules [70]. Mature DCs secrete IL-12 and IFN-α/-β as well as upregulate co-stimulating receptors such as CD80/86 to offer signals for CD8+ T cell differentiation and activation [71]. However, tumor cells in the TME can secrete inhibitory cytokines (IL-10, TGF, and RANKL) and decrease expression of costimulatory molecules to impede the maturation of DCs, converting it to a tolerance phenotype. The tolerogenic DCs produce IDO and IL-10, and express inducible co-stimulatory molecule ligand to stimulate the production of Tregs, finally inhibiting the immune response of CD8+ T cells [72].
TAMs
The exact role of tumor-associated macrophages (TAMs) in the tumor proximity depends on the macrophage phenotype conferred by TME [73]. Some TAMs play an active role in antigen presentation, while others, such as senescent TAMs, can suppress T-cell-mediated anti-tumor immune response, thus promoting tumor progression [74]. TAMs secrete immunosuppressive mediators such as IL-10 and TGF-β and downregulate receptors of Fas-L, PD-L1 and CD80/86, imposing negative impacts on CD8+ T cells, directly or indirectly [75]. It has been reported that TAMs can express CCL22, CCL17, and CCL18 to promote Tregs recruitment [76].
Neutrophils
Human neutrophils can be divided into two categories, namely N1 with anticancer effect and N2 with immunosuppressive effect [77]. N2 is an abnormal immature neutrophil or infiltrating mature neutrophils induced by TGF-β in TME [78]. Moreover, N2 neutrophils can induce the production of nitric oxide synthase, argininase 1 and reactive oxygen species, and prevent T cells from producing the active ingredient IFN-γ [79]. Besides, the high level of N2 neutrophil with expression of CCL2 and CCL17 could promote the aggregation of TAMs and Tregs and assist the maintenance of tumor immunosuppressive environment [80].
Other suppressive tactics
Cancer cells can evade immune surveillance while preserving immunogenicity and continue to stimulate the immune system, initiating a series of aberrant immune behaviors that further assist in tumor immune escape [81]. For instance, the constant stimulation of tumor antigens motivates T cells to work persistently, eventually reaching a state of exhaustion [64]. Canonically, the more severely depleted T cells are, the more inhibitory receptors are expressed on the cell surface, reducing the sensitivity to antigenic stimulation [82].
Solid malignancies are usually accompanied by anoxic acidic microenvironments [83]. Hypoxia-inducible factor-α (HIF-α) accumulates under these conditions and is associated with the initiation of multiple genes, especially key genes in angiogenesis and glycolytic pathway [84]. Cox-2/PGE2 pathway can enhance the activity of HIF2-α, activate the TGF/EGFR pathway to accelerate the nuclear transfer of HIF2-α, bind to the hypoxia binding region of vascular endothelial growth factor (VEGF) and cyclin D1 encoding gene promoter, thereby mediating the progression of lung cancer [85]. Hypoxic environment increases the expression of immunosuppressive receptor PD-L1 in tumor cells with HIF1-α present, while overexpressed PD-L1 interacts with programmed cell death protein-1 (PD-1) of CD8+ T cells to promote apoptosis of CD8+ T cells [86]. Furthermore, CD38 in hypoxic tumor environment takes partial responsibility for the formation of adenosine, while extracellular adenosine binds to adenosine receptors on CD8+ T cells, thus restraining CD8+ T cell activation and recruiting Tregs to further increase immune resistance in tumor cells [87].
Hypoxia-induced oncogene activation enhances glycolysis and lactic acid accumulation, leading to acidification in TME [84]. The pH-sensing protein mechanism allows tumors to survive in an acidic environment, while CD8+ effector and CD8+ memory T cells suffer from impaired function and shortened lifespan [88]. Thus, hypoxia-mediated extracellular acidification prevents T cells from expanding or performing their cytotoxic effects.
Cancer immunotherapy involving CD8+ T cells
Immune checkpoint therapy
Expressed on immune cells, immune checkpoints are a group of immunosuppressive molecules that can regulate the degree of immune activation and avoid autoimmune responses, so maintaining immune tolerance [89]. However, to change the fate of being eliminated, some tumor cells obviate such mechanism and deliver signal stimulation to immune cells, triggering T cell dysfunction and apoptosis [52]. Immunotherapy-based treatments, including immune checkpoint inhibitors (ICIs) typified by anti-PD-1/PD-L1 drugs, can re-activate immune cells by blocking immune checkpoints in cancer patients, so CD8+ T cytotoxicity against tumor cells can be restored [90].
In recent years, ICIs revolutionize the cancer treatment [91]. Research on monoclonal antibodies (mAbs) as inhibitors against the immune checkpoints that include PD-1, CTLA-4, TIGIT, LAG-3, and TIM-3, has undertaken a flurry of breakthroughs [92]. Among them, ipilimumab (anti-CTLA-4 mAb) is the first anti-cancer immunotherapy drug approved by the US FDA to treat unresectable or metastatic melanoma [93]. Since then, clinical development of antibody drugs based on immune checkpoint blockade has been upsurging. In 2015 and 2016, nivolumab (anti-PD-1 mAb) was respectively approved to treat metastatic melanoma and non-squamous non-small cell lung cancer (NSCLC), and in 2016 atezolizumab (anti-PD-L1 mAb) was greenlighted to treat metastatic NSCLC patients who had progressed after chemotherapy [94].
CD8+ T cell phenotype is a key player in anti-tumor immunity, orchestrating immunogenic cell death in cancers through several mechanisms (Fig. 3). Firstly, immunotherapy-activated CD8+ T cells trigger apoptotic cell death via release of perforin-granzyme or through Fas-Fas-L interaction. Upon encountering, CD8+ T cells perforate tumor cell membranes and unleash granzyme B into the cytoplasm, or alternatively Fas-expressed tumor cells are susceptible to be eliminated by Fas-L-enriched T cells, both initiating apoptosis [19]. Secondly, non-apoptotic cell death can be induced, directly or indirectly, by cytokine-secreting CD8+ T cells. Recent findings revealed that in head and neck squamous cell carcinoma blocking CTLA-4 can activate CD8+ T cells, releasing cytokines such as IFN-γ and TNF-α in the TME, further activating gasdermin intracellularly to induce pyroptosis, a programmed lytic cell death in tumors (Fig. 3) [95]. In parallel, IFN-γ released from activated CD8+ T cells during immunotherapy may suppress glutamate-cystine antiporter genes, so impairing cystine uptake by tumors. This impairment further promotes lipid peroxidation and accelerates ferroptosis in tumor cells [22].

The immunogenic cell death induced by activated CD8+ T cells. (A) CD8+ T cells induce apoptosis of tumor cells through granzyme B release. (B) CD8+ T cells induce apoptosis of tumor cells through death receptor ligand. CD8+ T cells induce (C) pyroptosis (D) or ferroptosis of tumor cells by secreting cytokines

Schematic representation of different adoptive T cell therapy modalities. In TIL-ACT therapy, tumor-resident T cells are isolated and rapidly expanded in vitro after surgery or biopsy. The same patient receives lymphodepletion, followed by infusion of expanded T cells back to the patient. In ACT with genetically modified T cells, T cells in the peripheral blood of patient are isolated and transduced by viral vectors to express specific TCR or CAR for next infusion of modified T cells into the patient. Reproduced with permission [157]
Despite groundbreaking success of ICIs, their overall response rates (ORRs) in patients remain low for most cancer types. Differences in ICI responses among individuals may come from various interplays between aberrant tumors and changing immune cells, where significant increases in immune cell populations, such as CD8+ T cells, after ICI treatment can be commonly observed [90, 96]. Accordingly, two major therapeutic strategies are proposed to improve the therapeutic responses in the clinical settings.
Firstly, identification of specific biomarkers in cancers may help predict the patient outcomes when treated using ICIs. Examining CD14+CD16b−HLA-DRhi in peripheral blood can increase the screening frequency of PD-1 mAb-sensitive stage IV melanoma patient [97]. Lately, breast cancer susceptibility gene 2 was found to be positively related to the therapeutic effect of PD-1 mAb [98]. Similarly, SMARCA4 mutations in patients with NSCLC were associated with improved survival after ICI therapy, suggesting that SMARCA4 detection may help assess the sensitivity of patients to immunotherapy [99]. Thus, timely and precise examination of predictive biomarkers in cancers may obviate non-responsive treatment of ICIs to some extent.
Secondly, synergistic combination of different ICIs or other therapeutics may increase the patient’s response rate and improve therapeutic benefits [100]. A Phase I clinical trial was conducted, where APX005M (anti-CD40 mAb), gemcitabine (DNA synthesis inhibitor) and nab-paclitaxel combined with nivolumab (anti-PD-1 mAb) were administered to treat patients with pancreatic cancer, demonstrating improved results [101]. Moreover, synergistic administration of atezolizumab (anti-PD-L1 mAb) and bevacizumab (anti-VEGF mAb) in treating unresectable hepatocellular carcinoma, currently approved by FDA, showed better tumor response than single administration, although mild-to-moderate adverse reactions were reported [102]. A Phase III clinical trial of atezolizumab combined with bevacizumab and paclitaxel in the treatment of NSCLC also demonstrated that compared with patients given bevacizumab and paclitaxel, the atezolizumab addition improved the survival rate of NSCLC patients [103]. Therefore, optimized combination of different therapy regimens may treat cancers more effectively, depending on enhanced understanding towards more results from real-world evidence studies.
Neoantigen vaccination
The tumor-specific vaccination that adopts cancerous antigens to activate the host immune system and so produce amplified and long-lasting anti-tumor responses, has become one powerful approach to prevent or suppress tumors [104]. Preventive vaccine lowers the risks of tumor occurrence by protecting the recipients from oncogenic factors due to viral infections, such as human papillomavirus vaccine, while therapeutic vaccine boosts patients’ immune responses and provokes memory immune cells to achieve long-term tumor remission [105]. Research on tumor vaccines focuses on tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). TAAs refer to molecules of 10-1000-fold increase in tumor cells compared to those in normal cells, such as HER2, p53, and MART-1, while TSAs are neoantigens such as NY-ESO-1 and CEA that are absent or restrictively expressed in normal cells, thus being preferred targets for tumor vaccines [106]. Neoantigens are mutated self-antigens in tumor cells, usually prioritized by whole-exome sequencing and RNA-seq of tumor samples from patients. But a shortage of targetable neoantigens in cancers set back the wide application of tumor vaccination and identified neoantigens induced low intratumoral T cell response, mostly due to heterogeneity in tumor burdens and immunosuppressive TME [107].
To solve those problems, personalized neoantigen vaccines were designed and applied to enhance immunotherapy. With multiple antigen epitopes for patients with glioblastoma, neoantigen-specific CD4+ and CD8+ T cells from the peripheral blood could infiltrate into intracranial glioblastoma in patients, paving a new avenue for immunotherapy targeting glioblastoma TME [108]. Similarly, peripheral blood mononuclear cells (PBMCs) were collected from individual patients with advanced lung cancers to derive DCs, which were pulsed by neoantigen peptides to obtain autologous DC vaccines for the personalized treatment. As a result, CD8+ T cells from PBMCs after vaccination had significantly higher secretion of IL-12 and stronger responses to mutant neoantigens than before vaccination [109]. Moreover, an acidity-responsive nanovaccine containing therapeutic reagents in the core and a model antigen on the surface was developed to greatly improve the antigen presentation by DCs and enhance drug delivery to tumors, shifting the immunosuppressive TME into a milieu in favor of antigen-specific CD4+ and CD8+ T cells [110].
Neoantigen vaccine may not stand alone to effectively diminish malignant tumors. Its combinations with other therapeutics, such as immune-enhanced adjuvants of granulocyte-macrophage colony-stimulating factor (GM-CSF) or ICIs, can enhance their anticancer effects on solid tumors [111]. For instance, a triple therapy combining neoantigen vaccine that induces the accumulation of CD8+ T cells, anti-PD-1 that suppresses immune tolerance signals, and agonist antibody against OX40 that induces T cell memory, was invented to treat mice bearing pancreatic adenocarcinoma of low immunogenicity and poor T cell infiltration, where neoantigen vaccine significantly increased tumor responsiveness [112]. Also, neoantigen DNA vaccination together with anti-PD-1 antibody mediated the colon cancer regression in a CD8+ T cell-dependent manner, as opposed to anti-PD-1 antibody alone that failed in tumor elimination [113]. Synthesized polymeric vesicles at nano scale co-delivered peptide antigens and stimulator of interferon genes (STING) agonists to promote DC maturation, eliciting inflammatory cytokine production, costimulatory marker expression, and antigen cross-presentation, leading to mobilization and activation of tumor-specific CD8+ T cells. This in return resulted in remarkable improvement in the response to anti-PD-1 and anti-CTLA-4 treatment in murine colorectal adenocarcinoma and melanoma models, respectively [114]. Thereby, personalized cancer vaccines, particularly in rational combination with ICIs, may represent a safe and efficient approach in various malignancies.
Cellular immunotherapy
Adoptive cell therapy (ACT) ushers cancer treatment into a new era and becomes a milestone in personalized medicine. T cell-based ACT depicts that a number of T cells are isolated from patients with various cancers and induced to transform and proliferate ex vivo, before these therapeutic cells gaining specificity for tumor cells are infused back to patients to improve tumor killing (Fig. 4) [115, 116]. Initially, the therapeutic cells used in ACT were sourced from the patient’s own TILs, but this method is limited by the low abundance of TILs and the inability to efficiently improve tumor dismantling [117]. Alternatively, T cells sourced from the peripheral blood of the patients are genetically modified with surface receptors that recognize tumor-specific antigens, cytokines, or signal transduction molecules [118].
Among them, CAR-T and TCR-T are the most developed immunotherapeutic strategies in the clinical settings. CAR-T cells express chimeric antigen receptor (CAR) molecules on the surface of T cells through gene editing. The process of recognizing tumors by CAR-T cells rely on the unique components of CAR rather than MHC molecule, so CAR-T cells can overcome the immune escape caused by the loss of MHC molecules to some extent [119]. Also genetically modified, TCR-T cells express tumor antigen-specific TCR on the surface of T cells. TCR-T cells can recognize a broad spectrum of antigen peptides presented by MHC molecules, including tumor cell surface antigens, intracellular antigens and neoantigens resulting from tumor mutations [120]. Given the fact that most proteins are expressed intracellularly and CAR-T cells only identify cell surface antigens, TCR-T thereby has a wider tumor antigen selectivity than CAR-T, whereas CAR-T cells are superior to TCR-T cells in terms of target affinity and therapeutic dosage [121]. Both regimens have shown remarkable effectiveness in treating hematological malignancies [122].
In contrast, ACT has achieved less success in treating solid tumors, commonly due to inadequate tumor infiltration and low T cell functionality and persistence [123]. The highly suppressive TME and antigen diversity in solid tumors often invalidate adoptive T cells and promote tumor immune escape, worsened by T cell exhaustion and off-target side effect, thwarting the therapeutic benefits. For CAR-T therapy, antigenic heterogeneity and antigen loss during treatment are two major problems, which may be simultaneously tackled by the strategies to target one surface-expressed antigen using CAR T-cells while also triggering endogenous T-cell responses against additional tumor antigens. In a recent study, vaccine-enhanced CAR-T cells effectively produced IFN-γ, improving the anti-cancer activity, and actively recruited and activated DCs in the tumors with simultaneous IL-12 secretion, triggering the antigen spreading to prevent antigen-negative tumor escape [124]. Through this enhancement, the endogenous immune system was primed, where robust CD4+ and CD8+ T-cell responses were gained against non-CAR tumor antigen, greatly increasing the tumor infiltration.
To strengthen CAR-T cell functionality and persistence, activation of TGF-β by repetitive challenges can epigenetically reprogram T cells toward a stem-like memory state and promote the robust expansion of human tissue-resident memory CD8+ CAR-T cells (CAR-TRM), attracting CAR-T cells to accumulate and eventually eliminate solid tumors [125]. Metabolic strategies were carried out to support TRM differentiation and durable function, and to facilitate tissue residency of memory CD8+ T cells in solid tumors for TIL enrichment, therefore maintaining their effector functions for improved prognosis [126]. However, it needs to be clarified that the phenotypic mixture of CD4+ and CD8+ T cells in CAR-T preparation has been long noted. As we here focus the role of CD8+ T cells in cancer immunotherapy, the anti-cancer efficacies of CD4+ T cells are by no means excluded. Similarly, CD4+ T cells can actively respond to mutant antigens, so mediating the tumor elimination [127]. In fact, patients with chronic lymphocytic leukemia, who were infused using CAR-T cells a decade ago and achieved complete cancer remission, exhibited highly activated CD4+ T cells that dominated the CAR-T cell population, corroborating a long-persisting CD4+ T cells [128].
As Claudin 18.2 (CLDN 18.2) is highly expressed in gastric cancers, humanized anti-CLDN18.2 antibodies were synthesized with specific binding affinity, and CLDN 18.2-expressing CAR-T cells were intravenously injected into mice bearing subcutaneous xenografts of gastric tumors. Consequently, elevated amounts of TNF-α, IL-2, and IFN-γ were produced by CAR-T cells treatment only in CLDN18.2-positive cancer cells, where predominant CD8+ T cells were evidenced, corroborating a persistent and highly tumor-infiltrating CAR-T therapy [129]. In this study on-target off-tumor toxicity as an adverse effect was not observed. In 2022, US FDA approved the investigational new drug application of CLDN 18.2-targeted CAR-T therapy in a Phase 1 clinical trial for the treatment of adult patients with relapsed or refractory gastric or pancreatic cancers, where insofar an acceptable safety profile was reported along with a mild cytokine release syndrome occurred in the majority of patients [130]. Two patients with metastatic pancreatic cancer who received CLDN 18.2 CAR-T cell therapy showed significantly increased amount of CD8+ T and Treg cells in peripheral blood, leading to well controlled tumor progression and reduced lung metastases [131]. Of note, heightened peak value and augmented copy number of CLDN 18.2 CAR were documented, representing successful expansion and persistence of functional CAR-T cells [130, 131].
For TCR-T cell therapy, most challenges come from off-tumor toxicity and tumor resistance. Off-tumor toxicity is generated by either on- or off-target detrimental effects, where on-target off-tumor toxicity is associated with antigen expression in normal tissues aside from those in cancers, and off-target off-tumor toxicity is related to the cross-reactivity of TCR by recognizing other antigens than designated one in normal cells [120]. These toxicities caused severe morbidity and even death in patients. The tumor-targeting specificity and precision of TCR can be improved by high-throughput screening and bioinformatics analysis to select the TCR of optimal affinity and avidity. Tumor resistance can be primary or secondary, whereas primary resistance mainly results from the antigen heterogeneity in tumors, and secondary resistance is acquired after TCR-T therapy onsets mostly due to the loss or attrition of MHC-I or upregulated immune checkpoints on tumor cells [132]. For instance, the interaction between TCR and PD-1 had a negative regulatory role in T cell antigen recognition [133, 134]. Thus, combined administration of genetically engineered T cells and ICIs may cope with those challenges to enhance the therapeutic efficacy of infused T cells [135].
Without genetic modification, ACT with TILs (TIL-ACT) first extracts the infiltrating lymphocytes from the patients’ tumor tissues through biopsy or surgery, and then expand them in vitro with IL-2 stimulation, followed by infusion of TILs back into the same patients for treatment. With cumulative experiences and technical advances on engineered ACT therapy, understanding towards maintaining T cell function and penetrating TME enables TIL-ACT to be actively applied in clinical studies. As TILs are naturally and originally occurring TME-infiltrated cells, they hold migration privilege back to the same tumor and possess minimal off-target toxicity, recycled by negative TCR selection [136]. Patients with metastatic tumors that previously had decent levels of intratumoral and stromal CD8+ TILs in a network with activated myeloid population can benefit from TIL-ACT treatment [137]. Research efforts have been continuously made to promote TILs manufacturing and standardize the therapy regimen to advance the efficacy of TIL-ACT and ensure optimal patient outcomes. Currently, TIL ACT has been gained FDA approval as the first-of-its-kind ACT therapy for solid tumors with other clinical trials ongoing.
Oncolytic virotherapy
Taking advantage of intrinsic lytic characteristics in naturally occurring viruses, oncolytic virotherapy is a triple interplay among the virus, the host immunity and the TME. Genetically modified oncolytic viruses (OVs) are equipped with exogenous materials to improve the infection specificity to cancer cells, promote the viral replication inside cancers, and ensure the tolerable biosafety, while keeping the normal cells minimally impacted and the tumor cells maximally lysated [138]. Talimogene laherparepvec (T-VEC) is the first approved oncolytic virotherapy for treatment of patients with unresectable or metastatic melanoma in 2015. T-VEC employs herpes simplex virus type 1 (HSV-1) with oncolytic properties but inactivates deleterious endogenous substances, such as neurotoxic factors, and inserts immunologically active boosters (e.g., GM-CSF encoding genes) to enhance further immune responses [139]. Through intratumoral injection, T-VEC disintegrates tumor cells and releases TAAs to provoke local or even distant anti-cancer immunity, recruiting immune cells to reverse TME and elevating CD8+ T cell responses. As a result, durable response rate and overall survival of patients with advanced melanoma were superior receiving T-VEC as first-line therapy [140]. Similarly, HSV-1-based Teserpaturev/G47Δ has been approved for the treatment of malignant glioma in Japan, and this therapy currently enters clinical trials of other solid malignancies, including prostate cancer and recurrent olfactory neuroblastoma [141].
Oncolytic virotherapies in combination with other treatments have been experimented in pre-clinical and clinical studies. Vesicular stomatitis virus (VSV), owing to its high sensitivity to type-I IFN inhibition and tropism to type-I IFN-deficient tumors, was overexpressed with IFN-β genes, which induced potent CD8+ T cell responses in murine models of subcutaneous B16 tumors and significant reduction of tumor volume. Compared to treatment using ICI alone, combinatorial treatment using VSV-based therapy together with ICI showed better anti-tumor efficacy with higher survival rates [142]. Furthermore, in lymphodepleted mice bearing B16 tumors, VSV-based therapy plus type-I IFN resistant CD8 CAR-T cells provided better tumor inhibition and higher survival rate, than type-I IFN resistant CD8 CAR-T cells alone or VSV plus wide-type CD8 CAR-T cells [143]. Thus, OV combined with ACT therapy improves the abundance of tumor reactive CD8+ T cells. Nevertheless, VSV-associated type-I IFN was also found to promote apoptosis in CD8 CAR-T cells where CAR was highly expressed, resulting in negative therapeutic effect [143]. It follows that OV fabrication is a multifactorial process, not automatically beneficial for combination immunotherapy. A recent Phase III study revealed that treatment using T-VEC plus pembrolizumab in immunotherapy-naïve patients with advanced melanoma, did not show improved progression-free survival or overall survival, compared to placebo-pembrolizumab treatment [144].
Nanomedicine
In the recent decade the combination of cancer nanomedicine and immunotherapy has attracted much attention. To improve the delivery of immunotherapeutic agents to tumor targets, nanotechnologies are implemented for enhancement of specific local tumor immune responses that are more reliable and durable with less systemic toxicity [145]. Three basic targeting strategies of nano-immunotherapy have been developed as follows (Fig. 5) [146].

Immunotherapy combined with nanomedicine. (A) Target tumor cells and induce immunogenic cell death. Nanomedicine encapsulating photosensitizers can target and enter tumor cells through enhanced permeability and retention effect of tumors, and then release therapeutic cargoes, triggering the translocation of calcium reticulum protein to the tumor cell membrane. The synergistic effect with reactive oxygen species produced by photosensitizers leads to cell death in tumors [147]. CRT, calcium reticulum protein; ROS, reactive oxygen species. (b) Target the TME. Nanomedicine coupled TLR agonist can induce DC maturation and prolong the duration of antigen presentation. In addition, the designed nanomedicine can affect the function of TAMs and manipulate TAMs to repolarize into M1 pro-inflammatory phenotype, producing TNF-α and iNOS [151]. iNOS, inducible nitric oxide synthase. (C) Target the peripheral immune system. Nanomedicine can encapsulate genes encoding CAR or TCR, which can selectively bind to CD8+ T cells in circulating blood, initiating receptor mediated endocytosis to internalize nanomedicine. With the release of mRNA from CD8+ T cells, T cells are reprogrammed to express CAR or TCR, inducing anti-tumor responses [152]. ECM, extracellular matrix
Firstly, tumor cells can be targeted to induce immunogenic cell death. A multimodal nanoparticle was constructed with coordination polymers containing Zn2+ and phosphate groups of an oxaliplatin prodrug in the core and coated with the photosensitizer pyrolipid conjugate [147]. In bilateral tumor-bearing mice of colon cancer model, intraperitoneal injection of nanoparticles induced an effective synergy between chemotherapy and photodynamic therapy upon light irradiation and activated immune response, triggering calreticulin relocation onto the tumor cell surface that summoned APC to process tumor antigens and activated antigen-specific CD8+ T cells. The nanoparticles, utilizing an enhanced permeability and retention effect to highly accumulate inside tumors, provide an immunogenic milieu in TME and a systemic tumor-specific immune response for the ensuing intraperitoneal injection of PD-L1 antibody, amplifying its antitumor efficacy [147]. By changing the photosensitizer moiety to a chemical drug, similar nanoparticles were fabricated and administered in combination of anti-PD-L1 antibody, greatly increasing the intratumoral infiltration of CD8+ T cells. As a result, this nanomedicine not only afforded complete tumor eradication, but also prevented tumor formation when mice of tumor remission were challenged again with cancer cells [148]. Therefore, this combination therapy may successfully prompt strong and long-lasting antitumor immunity.
Secondly, immune microenvironment can be targeted to revive tumor specific CD8+ T cells and increase their proliferation and infiltration. Nanoparticles based on functional peptide self-assembly and conjugated with Toll-like receptor (TLR7 and TLR8) agonist effectively induced DC maturation and extended the duration of antigen presentation, so potentiating neoantigen-specific stem-like CD8+ T cells to optimize the anti-tumor immunity [149]. In a murine lung cancer model, nanosized liposomes were loaded with dual inhibitors against both exosome biogenesis and release and coupled with antibodies against epithelial cell adhesion molecule, targeting and shifting cancer associated fibroblasts into quiescent fibroblasts. This TME reversal increased the cytotoxic T cell infiltration and augmented antitumor efficacy of PD-L1 antibody when administered synergistically [150]. Moreover, magnetic iron oxide nanoparticles with cationic polymer functionalization were internalized by myeloid-derived suppressor cells (MDSCs) (mainly TAMs) in brain tumor. Under radiation the nanoparticles repolarized MDSCs into M1 pro-inflammatory phenotype, producing a large amount of TNF-α and inducible nitric oxide synthase to relieve the suppression of CD8+ T cells in the inhibitory TME, which finally enhanced the anti-tumor efficacy [151].
Thirdly, the peripheral immune system can be targeted to reactivate and amplify CD8+ T cells. To this end, polymeric nanoparticles with surface conjugation to anti-CD8 antibody were loaded with mRNAs encoding CAR and TCR, which could selectively bind to CD8+ T cells in the circulating blood of murine models bearing human leukemia, prostate cancer, and hepatocellular carcinoma, to initiate receptor-mediated endocytosis. Following the mRNA unleashing in CD8+ T cells, T cells were reprogrammed to transiently express tumor-specific CAR or virus-specific TCR, which induced tumor regression comparable to therapeutic outcome of ACT [152]. Notably, tertiary lymphoid structure (TLS) is mainly formed by aggregation of immune cells such as CD8+ T and CD20+ B lymphocytes, which is observed in metastatic solid tumors and correlated to improved patient survival rate [153]. The presence of TLS was found in 94% of patients with high-grade ovarian cancer, and a strong co-occurrence of the infiltrated CD8+ T cells and B cell lineages was confirmed [154]. Recently, a nanovaccine made of Epstein-Barr virus nuclear antigen 1 (neoantigen) and a bi-adjuvant of Mn2+ (STING agonist for T cell activation) and cytosine-phosphate-guanine (TLR-9 agonist for B cell activation) was formulated and injected into mice bearing nasopharyngeal carcinoma. After administration, TLS was formed and normalized blood and lymph vessels were detected in tumor tissues, correlated with increased presence of CD8+ T lymphocytes in tumor and peripheral blood [155]. Thus, targeting TLS to promote the functional maturation of T and B cells and to obviate the adverse reactions can be a new direction to advance integration of nanomedicine and immunotherapy.
Conclusion
In this review we discussed the basics regarding the life cycle of CD8+ T cells, as well as how they develop into frontline warriors with robust anti-cancer activity. Simultaneously, we delved into the mechanism of tumor immune escape with recent research findings and deliberated the immunotherapy strategies in experimental and clinical medicine with new findings and ongoing challenges. Put together, CD8+ T cell plays an important role in anti-tumor immunity, as CD8+ T cell-based immunotherapies are becoming an indispensable component in the frontline cancer therapy. While continuous efforts towards the clinical applications of CD8+ T cell-based therapeutics are doubtlessly necessitated, the fundamental understanding of both tumor and T-cell biology, such as tumor heterogeneity and checkpoint blockade, to better design the immunotherapy regimen and to assess therapeutic outcome, is greatly warranted.
Data availability
Not applicable.
Abbreviations
- ACT:
-
adoptive cell therapy
- APCs:
-
antigen-presenting cells
- CAR-T:
-
chimeric antigen receptor T
- CAR-TRM :
-
tissue-resident memory CAR-T cells
- CTLA-4:
-
cytotoxic T lymphocyte antigen 4
- CLDN:
-
18.2-Claudin 18.2
- CXCR3:
-
chemokine receptor CXC motif 3
- DCs:
-
dendritic cells
- EVs:
-
extracellular vesicles
- FDA:
-
Food and Drug Administration
- HIF-α:
-
hypoxia-inducible factor-α
- HLA:
-
human leukocyte antigen
- HSV-1:
-
herpes simplex virus type 1
- IDO1:
-
indoleamine 2, 3-dioxygenase 1
- IFN-g:
-
interferon g
- LAG:
-
3-lymphocyte activation gene-3
- MDSCs:
-
myeloid-derived suppressor cells
- MHC:
-
major histocompatibility complex
- MHC I:
-
major histocompatibility complex class I
- NSCLC:
-
non-small cell lung cancer
- ORRs:
-
overall response rates
- Ovs:
-
oncolytic viruses
- OVV:
-
oncolytic virus vaccine
- PD-1:
-
programmed cell death protein-1
- PD-L1:
-
programmed cell death-ligand 1
- PDAC:
-
pancreatic ductal adenocarcinoma
- PBMCs:
-
peripheral blood mononuclear cells
- STING:
-
stimulator of interferon genes
- TAAs:
-
tumor-associated antigens
- TAMs:
-
tumor-associated macrophages
- TA-TLS:
-
tumor associated TLS
- TCR-T:
-
T-cell receptor T
- TGF-b:
-
transforming growth factor b
- TIGIT:
-
T cell immunoglobulin and ITIM domain
- TILs:
-
tumor-infiltrating lymphocytes
- TIM-3:
-
T cell immunoglobulin and mucin-domain containing-3
- TLS:
-
tertiary lymphoid structure
- TNF-α:
-
tumor necrosis factorα
- TME:
-
tumor microenvironment
- Tregs:
-
regulatory T cells
- TSAs:
-
tumor-specific antigens
- T-VEC:
-
talimogene laherparepvec
- VEGF:
-
vascular endothelial growth factor
References
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68.
CD8 < sup>+ T cells in the cancer-immunity cycle. Immunity 2023, 56(10):2231–2253.
Betof Warner A, Hamid O, Komanduri K, Amaria R, Butler MO, Haanen J, Nikiforow S, Puzanov I, Sarnaik A, Bishop MR et al. Expert consensus guidelines on management and best practices for tumor-infiltrating lymphocyte cell therapy. J Immunother Cancer 2024, 12(2).
Zhu M, Huang Y, Bender ME, Girard L, Kollipara R, Eglenen-Polat B, Naito Y, Savage TK, Huffman KE, Koyama S, et al. Evasion of Innate Immunity contributes to small cell Lung Cancer Progression and Metastasis. Cancer Res. 2021;81(7):1813–26.
Chen Y, Xu J, Wu X, Yao H, Yan Z, Guo T, Wang W, Wang P, Li Y, Yang X, et al. CD147 regulates antitumor CD8 + T-cell responses to facilitate tumor-immune escape. Cell Mol Immunol. 2021;18(8):1995–2009.
The Lancet O. CAR T-cell therapy for solid tumours. Lancet Oncol. 2021;22(7):893.
Vesely MD, Zhang T, Chen L. Resistance mechanisms to Anti-PD Cancer Immunotherapy. Annu Rev Immunol. 2022;40:45–74.
Simmons K, Kee BK, Raghav KPS, Johnson B, Kopetz S, Willis J, Dasari A, Sanchez EV, Ludford K, Parseghian CM, et al. Clinical outcomes following termination of immunotherapy due to long-term benefit in MSI-H colorectal cancer. J Clin Oncol. 2022;40(16suppl):3585–3585.
Siddiqui BA, Gheeya JS, Goswamy R, Bathala TK, Surasi DS, Gao J, Shah A, Campbell MT, Msaouel P, Goswami S et al. Durable responses in patients with genitourinary cancers following immune checkpoint therapy rechallenge after moderate-to-severe immune-related adverse events. J Immunother Cancer 2021, 9(7).
Allouchery M, Lombard T, Martin M, Rouby F, Sassier M, Bertin C, Atzenhoffer M, Miremont-Salame G, Perault-Pochat MC, Puyade M. Safety of immune checkpoint inhibitor rechallenge after discontinuation for grade ≥ 2 immune-related adverse events in patients with cancer. J Immunother Cancer 2020, 8(2).
Khan AB, Carpenter B, Santos ESP, Pospori C, Khorshed R, Griffin J, Velica P, Zech M, Ghorashian S, Forrest C, et al. Redirection to the bone marrow improves T cell persistence and antitumor functions. J Clin Invest. 2018;128(5):2010–24.
Ma J, Liu Y, Duan C, Wu S, Xie Y, Yang L, Li X, Wang Y, Zhang Y, Zhuang R. CD226 knockout reduces the development of CD8 + T by impairing the TCR sensitivity of double-positive thymocytes. Immunology. 2023;169(1):83–95.
Baumgaertner P, Schmidt J, Costa-Nunes CM, Bordry N, Guillaume P, Luescher I, Speiser DE, Rufer N, Hebeisen M. CD8 T cell function and cross-reactivity explored by stepwise increased peptide-HLA versus TCR affinity. Front Immunol. 2022;13:973986.
Schöllhorn A, Maia A, Kimmerle F, Born J, Rammensee HG, Dimitrov S, Gouttefangeas C. Staining of activated ß(2)-integrins in combination with CD137 and CD154 for sensitive identification of functional antigen-specific CD4(+) and CD8(+) T cells. Front Immunol. 2022;13:1107366.
Schmidt J, Chiffelle J, Perez MAS, Magnin M, Bobisse S, Arnaud M, Genolet R, Cesbron J, Barras D, Navarro Rodrigo B, et al. Neoantigen-specific CD8 T cells with high structural avidity preferentially reside in and eliminate tumors. Nat Commun. 2023;14(1):3188.
Watson NB, Patel RK, Kean C, Veazey J, Oyesola OO, Laniewski N, Grenier JK, Wang J, Tabilas C, Yee Mon KJ, et al. The gene regulatory basis of bystander activation in CD8 < sup>+ T cells. Sci Immunol. 2024;9(92):eadf8776.
Fu W, Lei C, Liu S, Cui Y, Wang C, Qian K, Li T, Shen Y, Fan X, Lin F, et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat Commun. 2019;10(1):4355.
Seo N, Shirakura Y, Tahara Y, Momose F, Harada N, Ikeda H, Akiyoshi K, Shiku H. Activated CD8 + T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat Commun. 2018;9(1):435.
Upadhyay R, Boiarsky JA, Pantsulaia G, Svensson-Arvelund J, Lin MJ, Wroblewska A, Bhalla S, Scholler N, Bot A, Rossi JM, et al. A critical role for Fas-mediated off-target tumor killing in T-cell immunotherapy. Cancer Discov. 2021;11(3):599–613.
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368(6494).
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579(7799):415–20.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4.
Huang D, Chen J, Yang L, Ouyang Q, Li J, Lao L, Zhao J, Liu J, Lu Y, Xing Y, et al. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat Immunol. 2018;19(10):1112–25.
He TS, Ji W, Zhang J, Lu J, Liu X. ALG-2 couples T cell activation and apoptosis by regulating proteasome activity and influencing MCL1 stability. Cell Death Dis. 2020;11(1):5.
Robinson GL, Dinsdale D, Macfarlane M, Cain K. Switching from aerobic glycolysis to oxidative phosphorylation modulates the sensitivity of mantle cell lymphoma cells to TRAIL. Oncogene. 2012;31(48):4996–5006.
Dubrot J, Du PP, Lane-Reticker SK, Kessler EA, Muscato AJ, Mehta A, Freeman SS, Allen PM, Olander KE, Ockerman KM, et al. In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat Immunol. 2022;23(10):1495–506.
Zeng X, Liao G, Li S, Liu H, Zhao X, Li S, Lei K, Zhu S, Chen Z, Zhao Y, et al. Eliminating METTL1-mediated accumulation of PMN-MDSCs prevents hepatocellular carcinoma recurrence after radiofrequency ablation. Hepatology. 2023;77(4):1122–38.
Carleton G, Lum JJ. Autophagy metabolically suppresses CD8(+) T cell antitumor immunity. Autophagy. 2019;15(9):1648–9.
MacNabb BW, Chen X, Tumuluru S, Godfrey J, Kasal DN, Yu J, Jongsma MLM, Spaapen RM, Kline DE, Kline J. Dendritic cells can prime anti-tumor CD8(+) T cell responses through major histocompatibility complex cross-dressing. Immunity. 2022;55(11):2206–8.
Cho H, Binder J, Weeratna R, Dermyer M, Dai S, Boccia A, Li W, Li S, Jooss K, Merson J, et al. Preclinical development of a vaccine-based immunotherapy regimen (VBIR) that induces potent and durable T cell responses to tumor-associated self-antigens. Cancer Immunol Immunother. 2023;72(2):287–300.
Lerner EC, Woroniecka KI, D’Anniballe VM, Wilkinson DS, Mohan AA, Lorrey SJ, Waibl-Polania J, Wachsmuth LP, Miggelbrink AM, Jackson JD, et al. CD8(+) T cells maintain killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL axis. Nat Cancer. 2023;4(9):1258–72.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. Allele-specific HLA loss and Immune escape in Lung Cancer Evolution. Cell. 2017;171(6):1259–e12711211.
Yajima T, Nishimura H, Wajjwalku W, Harada M, Kuwano H, Yoshikai Y. Overexpression of interleukin-15 in vivo enhances antitumor activity against MHC class I-negative and -positive malignant melanoma through augmented NK activity and cytotoxic T-cell response. Int J Cancer. 2002;99(4):573–8.
Cheng YC, Ku WC, Tseng TT, Wu CP, Li M, Lee SC. Anchorage independence altered vasculogenic phenotype of melanoma cells through downregulation in aminopeptidase N /syndecan-1/integrin β4 axis. Aging. 2020;12(17):16803–19.
Büll C, Boltje TJ, Balneger N, Weischer SM, Wassink M, van Gemst JJ, Bloemendal VR, Boon L, van der Vlag J, Heise T, et al. Sialic Acid Blockade suppresses Tumor Growth by enhancing T-cell–mediated tumor immunity. Cancer Res. 2018;78(13):3574.
Büll C, Boltje TJ, Balneger N, Weischer SM, Wassink M, van Gemst JJ, Bloemendal VR, Boon L, van der Vlag J, Heise T, et al. Sialic Acid Blockade suppresses Tumor Growth by enhancing T-cell-mediated tumor immunity. Cancer Res. 2018;78(13):3574–88.
Li X-Y, Moesta AK, Xiao C, Nakamura K, Casey M, Zhang H, Madore J, Lepletier A, Aguilera AR, Sundarrajan A, et al. Targeting CD39 in Cancer reveals an extracellular ATP- and inflammasome-driven tumor immunity. Cancer Discov. 2019;9(12):1754–73.
Li CY, Park HJ, Shin J, Baik JE, Mehrara BJ, Kataru RP. Tumor-Associated Lymphatics Upregulate MHC-II to suppress tumor-infiltrating lymphocytes. Int J Mol Sci 2022, 23(21).
Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–80.
Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, Blair T, Whiteford M, O’Brien D, Ahmad R, et al. TGFβ suppresses CD8 + T cell expression of CXCR3 and tumor trafficking. Nat Commun. 2020;11(1):1749.
Wu W, Wang W, Wang Y, Li W, Yu G, Li Z, Fang C, Shen Y, Sun Z, Han L, et al. IL-37b suppresses T cell priming by modulating dendritic cell maturation and cytokine production via dampening ERK/NF-κB/S6K signalings. Acta Biochim Biophys Sin (Shanghai). 2015;47(8):597–603.
Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, Fu K, Peng J, Wang Y, Ren Z, et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8 + T cell apoptosis. Cancer Cell. 2019;35(6):901–e915904.
Rivas JR, Liu Y, Alhakeem SS, Eckenrode JM, Marti F, Collard JP, Zhang Y, Shaaban KA, Muthusamy N, Hildebrandt GC, et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia. 2021;35(11):3188–200.
Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, Butler NS, Bruneau J, Shoukry NH, Krawczyk CM, et al. Interleukin-10 directly inhibits CD8(+) T cell function by enhancing N-Glycan branching to decrease Antigen Sensitivity. Immunity. 2018;48(2):299–e312295.
Oft M. Immune regulation and cytotoxic T cell activation of IL-10 agonists - preclinical and clinical experience. Semin Immunol. 2019;44:101325.
Zhang S, Wan J, Chen M, Cai D, Xu J, Chen Q. Tumor-infiltrating CD8 + T cells driven by the Immune Checkpoint-Associated Gene IDO1 are Associated with Cervical Cancer Prognosis. Front Oncol. 2021;11:720447.
Tang K, Wu YH, Song Y, Yu B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol. 2021;14(1):68.
Huang Q, Xia J, Wang L, Wang X, Ma X, Deng Q, Lu Y, Kumar M, Zhou Z, Li L, et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol. 2018;11(1):58.
Blair AB, Kleponis J, Thomas DL 2nd, Muth ST, Murphy AG, Kim V, Zheng L. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J Clin Invest. 2019;129(4):1742–55.
Lou Q, Liu R, Yang X, Li W, Huang L, Wei L, Tan H, Xiang N, Chan K, Chen J, et al. miR-448 targets IDO1 and regulates CD8(+) T cell response in human colon cancer. J Immunother Cancer. 2019;7(1):210–210.
Simons KH, de Vries MR, Peters HAB, Jukema JW, Quax PHA, Arens R. CD8 + T cells protect during Vein Graft Disease Development. Front Cardiovasc Med. 2019;6:77.
Zhu J, Petit PF, Van den Eynde BJ. Apoptosis of tumor-infiltrating T lymphocytes: a new immune checkpoint mechanism. Cancer Immunol Immunother. 2019;68(5):835–47.
Chang H, Jung WY, Kang Y, Lee H, Kim A, Kim HK, Shin BK, Kim BH. Programmed death-ligand 1 expression in gastric adenocarcinoma is a poor prognostic factor in a high CD8 + tumor infiltrating lymphocytes group. Oncotarget. 2016;7(49):80426–34.
Zhao D, Zhai B, He C, Tan G, Jiang X, Pan S, Dong X, Wei Z, Ma L, Qiao H, et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014;26(5):1030–9.
Sheng YH, Giri R, Davies J, Schreiber V, Alabbas S, Movva R, He Y, Wu A, Hooper J, McWhinney B, et al. A Nucleotide Analog prevents colitis-Associated Cancer via Beta-Catenin independently of inflammation and autophagy. Cell Mol Gastroenterol Hepatol. 2021;11(1):33–53.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5.
Wang Z, Li B, Zhou L, Yu S, Su Z, Song J, Sun Q, Sha O, Wang X, Jiang W, et al. Prodigiosin inhibits Wnt/β-catenin signaling and exerts anticancer activity in breast cancer cells. Proc Natl Acad Sci U S A. 2016;113(46):13150–5.
Peng M, Ren J, Jing Y, Jiang X, Xiao Q, Huang J, Tao Y, Lei L, Wang X, Yang Z, et al. Tumour-derived small extracellular vesicles suppress CD8 + T cell immune function by inhibiting SLC6A8-mediated creatine import in NPM1-mutated acute myeloid leukaemia. J Extracell Vesicles. 2021;10(13):e12168.
Hu S, Ma J, Su C, Chen Y, Shu Y, Qi Z, Zhang B, Shi G, Zhang Y, Zhang Y, et al. Engineered exosome-like nanovesicles suppress tumor growth by reprogramming tumor microenvironment and promoting tumor ferroptosis. Acta Biomater. 2021;135:567–81.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560(7718):382–6.
Strnadová K, Pfeiferová L, Přikryl P, Dvořánková B, Vlčák E, Frýdlová J, Vokurka M, Novotný J, Šáchová J, Hradilová M, et al. Exosomes produced by melanoma cells significantly influence the biological properties of normal and cancer-associated fibroblasts. Histochem Cell Biol. 2022;157(2):153–72.
Zhou J, Yang Y, Wang W, Zhang Y, Chen Z, Hao C, Zhang J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4 + T cells through their microRNA cargo. Exp Cell Res. 2018;371(2):364–71.
Shi L, Feng M, Du S, Wei X, Song H, Yixin X, Song J, Wenxian G. Adenosine Generated by Regulatory T Cells induces CD8(+) T cell exhaustion in gastric Cancer through A2aR pathway. Biomed Res Int. 2019;2019:4093214–4093214.
Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Investig. 2016;126(4):1216–23.
Sorrentino C, Hossain F, Rodriguez PC, Sierra RA, Pannuti A, Osborne BA, Minter LM, Miele L, Morello S. Adenosine A2A receptor stimulation inhibits TCR-Induced Notch1 activation in CD8 + T-Cells. Front Immunol. 2019;10:162.
Byrnes JR, Weeks AM, Shifrut E, Carnevale J, Kirkemo L, Ashworth A, Marson A, Wells JA. Hypoxia is a Dominant Remodeler of the Effector T Cell Surface Proteome Relative To Activation and Regulatory T Cell Suppression. Mol Cell Proteom. 2022;21(4):100217.
Korbecki J, Kojder K, Barczak K, Simińska D, Gutowska I, Chlubek D, Baranowska-Bosiacka I. Hypoxia alters the expression of CC chemokines and CC Chemokine Receptors in a Tumor-A Literature Review. Int J Mol Sci. 2020;21(16):5647.
Najafi M, Farhood B, Mortezaee K. Contribution of regulatory T cells to cancer: a review. J Cell Physiol. 2019;234(6):7983–93.
González-Navajas JM, Fan DD, Yang S, Yang FM, Lozano-Ruiz B, Shen L, Lee J. The impact of Tregs on the Anticancer immunity and the efficacy of Immune checkpoint inhibitor therapies. Front Immunol. 2021;12:625783.
Barends M, Koller N, Schölz C, Durán V, Bošnjak B, Becker J, Döring M, Blees H, Förster R, Kalinke U, et al. Dynamic interactome of the MHC I peptide loading complex in human dendritic cells. Proc Natl Acad Sci U S A. 2023;120(25):e2219790120.
Sköld AE, Mathan TSM, van Beek JJP, Flórez-Grau G, van den Beukel MD, Sittig SP, Wimmers F, Bakdash G, Schreibelt G, de Vries IJM. Naturally produced type I IFNs enhance human myeloid dendritic cell maturation and IL-12p70 production and mediate elevated effector functions in innate and adaptive immune cells. Cancer Immunol Immunother. 2018;67(9):1425–36.
Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, Bachelot T, Treilleux I, Goddard-Leon S, Lavergne E, et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4 + T cells. Cancer Res. 2012;72(23):6130–41.
Li S, Yu J, Huber A, Kryczek I, Wang Z, Jiang L, Li X, Du W, Li G, Wei S, et al. Metabolism drives macrophage heterogeneity in the tumor microenvironment. Cell Rep. 2022;39(1):110609.
Wang L, He T, Liu J, Tai J, Wang B, Chen Z, Quan Z. Pan-cancer analysis reveals tumor-associated macrophage communication in the tumor microenvironment. Exp Hematol Oncol. 2021;10(1):31.
Burstyn-Cohen T, Maimon A. TAM receptors, phosphatidylserine, inflammation, and Cancer. Cell Commun Signal. 2019;17(1):156–156.
Huang L, Zhao Y, Shan M, Wang S, Chen J, Liu Z, Xu Q. Targeting crosstalk of STAT3 between tumor-associated M2 macrophages and Tregs in colorectal cancer. Cancer Biol Ther. 2023;24(1):2226418.
Chen Q, Yin H, Liu S, Shoucair S, Ding N, Ji Y, Zhang J, Wang D, Kuang T, Xu X et al. Prognostic value of tumor-associated N1/N2 neutrophil plasticity in patients following radical resection of pancreas ductal adenocarcinoma. J Immunother Cancer 2022, 10(12).
Chung JY, Tang PC, Chan MK, Xue VW, Huang XR, Ng CS, Zhang D, Leung KT, Wong CK, Lee TL, et al. Smad3 is essential for polarization of tumor-associated neutrophils in non-small cell lung carcinoma. Nat Commun. 2023;14(1):1794.
Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang Z, Wang C, Zhang Z, Xia W, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40(5):785–800.
Hu J, Zhang L, Xia H, Yan Y, Zhu X, Sun F, Sun L, Li S, Li D, Wang J, et al. Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell lung cancer revealed by single-cell RNA sequencing. Genome Med. 2023;15(1):14.
Du WQ, Zhu ZM, Jiang X, Kang MJ, Pei DS. COPS6 promotes tumor progression and reduces CD8(+) T cell infiltration by repressing IL-6 production to facilitate tumor immune evasion in breast cancer. Acta Pharmacol Sin. 2023;44(9):1890–905.
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–710.
Ning WR, Jiang D, Liu XC, Huang YF, Peng ZP, Jiang ZZ, Kang T, Zhuang SM, Wu Y, Zheng L. Carbonic anhydrase XII mediates the survival and prometastatic functions of macrophages in human hepatocellular carcinoma. J Clin Invest 2022, 132(7).
Ferreira BL, Leite GGF, Brunialti MKC, Assuncao M, Azevedo LCP, Freitas F, Salomao R. HIF-1α and Hypoxia Responsive genes are differentially expressed in Leukocytes from survivors and non-survivors patients during Clinical Sepsis. Shock. 2021;56(1):80–91.
Dong X-F, Liu T-Q, Zhi X-T, Zou J, Zhong J-T, Li T, Mo X-L, Zhou W, Guo W-W, Liu X, et al. COX-2/PGE2 Axis regulates HIF2α activity to promote Hepatocellular Carcinoma Hypoxic Response and reduce the sensitivity of Sorafenib Treatment. Clin Cancer Res. 2018;24(13):3204.
Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in Cancer cells. Cancer Res. 2014;74(3):665.
Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, Villalobos PA, Cascone T, Liu X, Tan L, et al. CD38-Mediated immunosuppression as a mechanism of Tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 2018;8(9):1156–75.
Chen X, Jaiswal A, Costliow Z, Herbst P, Creasey EA, Oshiro-Rapley N, Daly MJ, Carey KL, Graham DB, Xavier RJ. pH sensing controls tissue inflammation by modulating cellular metabolism and endo-lysosomal function of immune cells. Nat Immunol. 2022;23(7):1063–75.
Meng L, Wu H, Wu J, Ding P, He J, Sang M, Liu L. Mechanisms of immune checkpoint inhibitors: insights into the regulation of circular RNAS involved in cancer hallmarks. Cell Death Dis. 2024;15(1):3.
Castiglioni A, Yang Y, Williams K, Gogineni A, Lane RS, Wang AW, Shyer JA, Zhang Z, Mittman S, Gutierrez A, et al. Combined PD-L1/TGFβ blockade allows expansion and differentiation of stem cell-like CD8 T cells in immune excluded tumors. Nat Commun. 2023;14(1):4703.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905.
Velcheti V. K Schalper 2016 Basic overview of current immunotherapy approaches in Cancer. Am Soc Clin Oncol Educational Book 36 298–308.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with Ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Wang S, Wu ZZ, Zhu SW, Wan SC, Zhang MJ, Zhang BX, Yang QC, Xiao Y, Li H, Mao L, et al. CTLA-4 blockade induces tumor pyroptosis via CD8(+) T cells in head and neck squamous cell carcinoma. Mol Ther. 2023;31(7):2154–68.
Budhu A, Pehrsson EC, He A, Goyal L, Kelley RK, Dang H, Xie C, Monge C, Tandon M, Ma L, et al. Tumor biology and immune infiltration define primary liver cancer subsets linked to overall survival after immunotherapy. Cell Rep Med. 2023;4(6):101052.
Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, Becher B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24(2):144–53.
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35–44.
Schoenfeld AJ, Bandlamudi C, Lavery JA, Montecalvo J, Namakydoust A, Rizvi H, Egger J, Concepcion CP, Paul S, Arcila ME, et al. The genomic Landscape of SMARCA4 alterations and associations with outcomes in patients with Lung Cancer. Clin Cancer Res. 2020;26(21):5701–8.
Lv Z, Zhang P, Li D, Qin M, Nie L, Wang X, Ai L, Feng Z, Odhiambo WO, Ma Y, et al. CD19-targeting fusion protein combined with PD1 antibody enhances anti-tumor immunity in mouse models. Oncoimmunology. 2020;9(1):1747688.
O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, Fisher G, Rahma O, Lyman JP, Cabanski CR, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021;22(1):118–31.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. Long-term outcomes with Nivolumab Plus Ipilimumab or Nivolumab alone Versus Ipilimumab in patients with Advanced Melanoma. J Clin Oncol. 2022;40(2):127–37.
Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D, et al. Atezolizumab plus Bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respiratory Med. 2019;7(5):387–401.
Medikonda R, Pant A, Lim M. Immunotherapy as a New Therapeutic Approach for Brain and spinal cord tumors. Adv Exp Med Biol. 2023;1394:73–84.
Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15(1):28.
Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–22.
Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discovery. 2022;21(4):261–82.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. Neoantigen vaccine generates intratumoral T cell responses in phase ib glioblastoma trial. Nature. 2019;565(7738):234–9.
Ding Z, Li Q, Zhang R, Xie L, Shu Y, Gao S, Wang P, Su X, Qin Y, Wang Y, et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct Target Therapy. 2021;6(1):26.
Xie X, Feng Y, Zhang H, Su Q, Song T, Yang G, Li N, Wei X, Li T, Qin X, et al. Remodeling tumor immunosuppressive microenvironment via a novel bioactive nanovaccines potentiates the efficacy of cancer immunotherapy. Bioact Mater. 2022;16:107–19.
Li B, Jing P, Zheng G, Pi C, Zhang L, Yin Z, Xu L, Qiu J, Gu H, Qiu T, et al. Neo-intline: integrated pipeline enables neoantigen design through the in-silico presentation of T-cell epitope. Signal Transduct Target Therapy. 2023;8(1):397.
Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, Woolman S, Vithayathil T, Glickman LH, Ndubaku CO et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3(20).
Tondini E, Arakelian T, Oosterhuis K, Camps M, van Duikeren S, Han W, Arens R, Zondag G, van Bergen J, Ossendorp F. A poly-neoantigen DNA vaccine synergizes with PD-1 blockade to induce T cell-mediated tumor control. Oncoimmunology. 2019;8(11):1652539.
Shae D, Baljon JJ, Wehbe M, Christov PP, Becker KW, Kumar A, Suryadevara N, Carson CS, Palmer CR, Knight FC, et al. Co-delivery of peptide neoantigens and stimulator of Interferon genes agonists enhances response to Cancer vaccines. ACS Nano. 2020;14(8):9904–16.
Du S, Yan J, Xue Y, Zhong Y, Dong Y. Adoptive cell therapy for cancer treatment. Explor (Beijing). 2023;3(4):20210058.
Olson DJ, Odunsi K. Adoptive cell therapy for nonhematologic solid tumors. J Clin Oncol. 2023;41(18):3397–407.
Karbach J, Kiselicki D, Brand K, Wahle C, Sinelnikov E, Gustavus D, Hoffmeister H, Prisack HB, Atmaca A, Jäger E. Tumor-infiltrating lymphocytes mediate complete and durable remission in a patient with NY-ESO-1 expressing prostate cancer. J Immunother Cancer 2023, 11(1).
Leon E, Ranganathan R, Savoldo B. Adoptive T cell therapy: boosting the immune system to fight cancer. Semin Immunol. 2020;49:101437.
Albelda SM. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat Rev Clin Oncol. 2024;21(1):47–66.
Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: state of the art and perspectives. Sci Adv. 2023;9(7):eadf3700.
Yang D, Duan Z, Yuan P, Ding C, Dai X, Chen G, Wu D. How does TCR-T cell therapy exhibit a superior anti-tumor efficacy. Biochem Biophys Res Commun. 2023;687:149209.
Li D, Li X, Zhou W-L, Huang Y, Liang X, Jiang L, Yang X, Sun J, Li Z, Han W-D, et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct Target Therapy. 2019;4(1):35.
Irvine DJ, Maus MV, Mooney DJ, Wong WW. The future of engineered immune cell therapies. Science. 2022;378(6622):853–8.
Ma L, Hostetler A, Morgan DM, Maiorino L, Sulkaj I, Whittaker CA, Neeser A, Pires IS, Yousefpour P, Gregory J, et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell. 2023;186(15):3148–e31653120.
Jung IY, Noguera-Ortega E, Bartoszek R, Collins SM, Williams E, Davis M, Jadlowsky JK, Plesa G, Siegel DL, Chew A, et al. Tissue-resident memory CAR T cells with stem-like characteristics display enhanced efficacy against solid and liquid tumors. Cell Rep Med. 2023;4(6):101053.
Reina-Campos M, Heeg M, Kennewick K, Mathews IT, Galletti G, Luna V, Nguyen Q, Huang H, Milner JJ, Hu KH, et al. Metabolic programs of T cell tissue residency empower tumour immunity. Nature. 2023;621(7977):179–87.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4 + T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5.
Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, Gao P, Bandyopadhyay S, Sun H, Zhao Z, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. 2022;602(7897):503–9.
Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G, Zhang H, Shi B, Jia J, Li Q, et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the treatment of gastric Cancer. J Natl Cancer Inst. 2019;111(4):409–18.
Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, Zhang M, Peng Z, Zhou J, Cao Y, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. 2022;28(6):1189–98.
Qi C, Xie T, Zhou J, Wang X, Gong J, Zhang X, Li J, Yuan J, Liu C, Shen L. CT041 CAR T cell therapy for Claudin18.2-positive metastatic pancreatic cancer. J Hematol Oncol. 2023;16(1):102.
Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. 2019;290(1):127–47.
Li K, Yuan Z, Lyu J, Ahn E, Davis SJ, Ahmed R, Zhu C. PD-1 suppresses TCR-CD8 cooperativity during T-cell antigen recognition. Nat Commun. 2021;12(1):2746.
Chan W, Cao YM, Zhao X, Schrom EC, Jia D, Song J, Sibener LV, Dong S, Fernandes RA, Bradfield CJ et al. TCR ligand potency differentially impacts PD-1 inhibitory effects on diverse signaling pathways. J Exp Med 2023, 220(12).
Rossetti R, Brand H, Lima SCG, Furtado IP, Silveira RM, Fantacini DMC, Covas DT, de Souza LEB. Combination of genetically engineered T cells and immune checkpoint blockade for the treatment of cancer. Immunotherapy Adv 2022, 2(1).
Wang S, Sun J, Chen K, Ma P, Lei Q, Xing S, Cao Z, Sun S, Yu Z, Liu Y, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19(1):140.
Barras D, Ghisoni E, Chiffelle J, Orcurto A, Dagher J, Fahr N, Benedetti F, Crespo I, Grimm AJ, Morotti M, et al. Response to tumor-infiltrating lymphocyte adoptive therapy is associated with preexisting CD8 < sup>+ T-myeloid cell networks in melanoma. Sci Immunol. 2024;9(92):eadg7995.
Lin D, Shen Y, Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduct Target Therapy. 2023;8(1):156.
Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20(3):160–77.
Johnson DB, Puzanov I, Kelley MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7(6):611–9.
Frampton JE. Teserpaturev/G47∆: first approval. BioDrugs. 2022;36(5):667–72.
Kottke T, Tonne J, Evgin L, Driscoll CB, van Vloten J, Jennings VA, Huff AL, Zell B, Thompson JM, Wongthida P, et al. Oncolytic virotherapy induced CSDE1 neo-antigenesis restricts VSV replication but can be targeted by immunotherapy. Nat Commun. 2021;12(1):1930.
Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, Schuelke M, Ayasoufi K, Driscoll CB, Shim KG, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun. 2020;11(1):3187.
Chesney JA, Ribas A, Long GV, Kirkwood JM, Dummer R, Puzanov I, Hoeller C, Gajewski TF, Gutzmer R, Rutkowski P et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined With Pembrolizumab for Advanced Melanoma. Journal of Clinical Oncology 2023, 41(3):528–540.
Musetti S, Huang L. Nanoparticle-mediated remodeling of the Tumor Microenvironment to Enhance Immunotherapy. ACS Nano. 2018;12(12):11740–55.
Sun Q, Bai X, Sofias AM, van der Meel R, Ruiz-Hernandez E, Storm G, Hennink WE, De Geest B, Kiessling F, Yu H-, et al. Cancer nanomedicine meets immunotherapy: opportunities and challenges. Acta Pharmacol Sin. 2020;41(7):954–8.
He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR, Lin W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat Commun. 2016;7:12499.
Duan X, Chan C, Han W, Guo N, Weichselbaum RR, Lin W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat Commun. 2019;10(1):1899.
Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mulè MP, et al. Intravenous nanoparticle vaccination generates stem-like TCF1(+) neoantigen-specific CD8(+) T cells. Nat Immunol. 2021;22(1):41–52.
Freag MS, Mohammed MT, Kulkarni A, Emam HE, Maremanda KP, Elzoghby AO. Modulating tumoral exosomes and fibroblast phenotype using nanoliposomes augments cancer immunotherapy. Sci Adv. 2024;10(9):eadk3074.
Wu C, Muroski ME, Miska J, Lee-Chang C, Shen Y, Rashidi A, Zhang P, Xiao T, Han Y, Lopez-Rosas A, et al. Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment. Nanomedicine. 2019;16:126–37.
Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. 2020;11(1):6080.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–5.
Ukita M, Hamanishi J, Yoshitomi H, Yamanoi K, Takamatsu S, Ueda A, Suzuki H, Hosoe Y, Furutake Y, Taki M et al. CXCL13-producing CD4 + T cells accumulate in the early phase of tertiary lymphoid structures in ovarian cancer. JCI Insight 2022, 7(12).
Wen Z, Liu H, Qiao D, Chen H, Li L, Yang Z, Zhu C, Zeng Z, Chen Y, Liu L. Nanovaccines fostering Tertiary Lymphoid structure to Attack Mimicry Nasopharyngeal Carcinoma. ACS Nano. 2023;17(8):7194–206.
Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72(13):3125–30.
Rohaan MW, Wilgenhof S, Haanen J. Adoptive cellular therapies: the current landscape. Virchows Arch. 2019;474(4):449–61.
Acknowledgements
We thank the financial support from Jiangsu University.
Funding
This work was supported by the Nature Science Foundation of Jiangsu Province [grant number BK20230528]; the National Natural Science Foundation of China [grant number 82303767].
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript writing and agreed on the manuscript submission and publication.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Yu, D., Qian, H. et al. CD8+ T cell-based cancer immunotherapy. J Transl Med 22, 394 (2024). https://doi.org/10.1186/s12967-024-05134-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05134-6