
Abstract
Background
Effective bone formation relies on osteoblast differentiation, a process subject to intricate post-translational regulation. Ubiquitin-specific proteases (USPs) repress protein degradation mediated by the ubiquitin-proteasome pathway. Several USPs have been documented to regulate osteoblast differentiation, but whether other USPs are involved in this process remains elusive.
Methods
In this study, we conducted a comparative analysis of 48 USPs in differentiated and undifferentiated hFOB1.19 osteoblasts, identifying significantly upregulated USPs. Subsequently, we generated USP knockdown hFOB1.19 cells and evaluated their osteogenic differentiation using Alizarin red staining. We also assessed cell viability, cell cycle progression, and apoptosis through MTT, 7-aminoactinomycin D staining, and Annexin V/PI staining assays, respectively. Quantitative PCR and Western blotting were employed to measure the expression levels of osteogenic differentiation markers. Additionally, we investigated the interaction between the USP and its target protein using co-immunoprecipitation (co-IP). Furthermore, we depleted the USP in hFOB1.19 cells to examine its effect on the ubiquitination and stability of the target protein using immunoprecipitation (IP) and Western blotting. Finally, we overexpressed the target protein in USP-deficient hFOB1.19 cells and evaluated its impact on their osteogenic differentiation using Alizarin red staining.
Results
USP36 is the most markedly upregulated USP in differentiated hFOB1.19 osteoblasts. Knockdown of USP36 leads to reduced viability, cell cycle arrest, heightened apoptosis, and impaired osteogenic differentiation in hFOB1.19 cells. USP36 interacts with WD repeat-containing protein 5 (WDR5), and the knockdown of USP36 causes an increased level of WDR5 ubiquitination and accelerated degradation of WDR5. Excessive WDR5 improved the impaired osteogenic differentiation of USP36-deficient hFOB1.19 cells.
Conclusions
These observations suggested that USP36 may function as a key regulator of osteoblast differentiation, and its regulatory mechanism may be related to the stabilization of WDR5.
Similar content being viewed by others


Introduction
Osteoblasts are bone-forming cells that synthesize and secrete bone matrix, playing an essential role in bone formation, remodeling, and healing [1]. Dysfunction of osteoblasts is responsible for multiple types of diseases including osteoporosis, skeletal dysplasia, and primary bone cancers [2]. Consequently, enhancing osteoblast function holds promise for promoting bone regeneration.
The differentiation of osteoblasts is a multiple-step process controlled by various genes and signaling pathways [3]. Moreover, the key players for osteoblast differentiation themselves are also tightly regulated at transcriptional and post-translational levels. For instance, the expression of RUNX2, a key transcription factor governing osteoblast commitment and differentiation, is regulated by multiple signaling pathways such as WNT, BMP, HH, and PTH pathways [4,5,6,7]. Moreover, the expression of c-MYC, another essential transcription factor that controls osteoblast differentiation [8], is targeted by the WNT signaling [9]. While the mechanisms underlying the regulation of these transcription factors have been intensively investigated, their post-translational level regulation during osteoblast differentiation received less attention.
Ubiquitination is an essential cellular process for protein degradation, which is accomplished by a cascade of enzymes known as E1-E2-E3 [10]. E3 ubiquitin ligases, a class of enzymes within this cascade, are responsible for selecting target proteins for ubiquitin conjugation, which can be removed by deubiquitinating enzymes, including ubiquitin-specific proteases (USPs) [11, 12]. Emerging research has highlighted the involvement of USPs in the regulation of osteoblast differentiation. For example, USP4 inhibits the differentiation of osteoblasts by stabilizing Dishevelled, thereby reducing the canonical WNT pathway [13]. By contrast, USP15 promotes bone formation by deubiquitinating β-catenin in osteoblasts [14]. Moreover, USP34 facilitates osteoblast differentiation by regulating the BMP2 signaling [15]. In addition, USP7 can either positively regulate osteoblast differentiation by stabilizing RUNX2 [16], or exert an opposite function by stabilizing AXIN protein, which inhibits the canonical WNT signaling [17]. Nevertheless, the precise roles of other USPs in osteoblast differentiation remain largely unknown.
In this study, we aim to identify additional USPs that may influence osteoblast differentiation and delineate the underlying mechanism. To accomplish this, we initially compared the expression profiles of USPs in differentiated and undifferentiated hFOB1.19 osteoblasts, identifying USP36 as the most upregulated USP in these cells. Subsequently, we depleted USP36 in hFOB1.19 cells and evaluated their osteogenic differentiation, viability, cell cycle, and apoptosis. We then predicted the substrate protein of USP36 and investigated their interaction in hFOB1.19 cells. Finally, we overexpressed this protein in USP36-deficient hFOB1.19 cells to determine if it could rescue the mitigated osteogenic differentiation of these cells.
Materials and methods
Cell lines
hFOB1.19 and HEK293T cells were purchased from Immocell Biotechnology (Xiamen, China) and maintained in DMEM/F12 medium (IMC-205, Immocell Biotechnology) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) and 100U/mL penicillin-streptomycin in incubators at 33.5–37 °C, respectively, with 5% CO2. Osteogenic differentiation was induced by treatment of the following osteogenic medium: DMEM/F12 medium supplemented with 10% fetal bovine serum, 2 mM L-Glutamine (25030-149, Gibco), 0.1 µM dexamethasone (D4902-25MG, Sigma-Aldrich, Burlington, MA, USA), 10 mM Beta-glycerophosphate disodium salt hydrate (G9422-10G, Sigma-Aldrich), and 50 µg/mL L-ascorbic acid (A4034-100G, Sigma-Aldrich).
Construction of plasmid
The USP36-targeting short hairpin RNA or scrambled control was inserted into PLKO.1-TRC-puro vector, and the resulting plasmids were named shUSP36 and shNC, respectively. Subsequently, the plasmids were sequenced to confirm the accuracy of the inserted sequences. The primer sequences are shown in Table 1.
Lentiviral transfection
To prepare the lentiviral particles, 9 µg of shUSP36 or shNC and the suitable packaging plasmids (3 µg of pMD2G and 6 µg of pspax2) were co-transfected into 1.4 × 107 HEK293T cells using Lipofectamine® 2000 (Invitrogen, Waltham, MA, USA). After 48 h, the virus-containing supernatant was collected for concentration, and the virus titer was determined as described previously [18]. Subsequently, the lentivirus was incubated with hFOB1.19 cells at a multiplicity of infection (MOI) of 10, in the presence of 8 µg/ml polybrene (107,689, Sigma-Aldrich). After another 48 h of culture, the medium was replaced by the freshly prepared culture medium containing 1.0 µg/ml puromycin (P9620, Sigma-Aldrich). After another 72 h of culture, the cells were subject to further analysis.
Alizarin red staining
hFOB1.19 cells cultured with the osteogenic medium for 21 days were gently washed with phosphate-buffered solution (PBS) and incubated with 4% paraformaldehyde for 30 min at room temperature (RT), followed by another wash of PBS. After incubating with Alizarin Red S Solution (ALIR-10,001, OriCell, Shanghai, China) for 15 min, the cells were gently washed with PBS again and subject to observation and imaging using an optical microscope.
MTT assay
MTT assay was conducted to evaluate the cell viability. Briefly, control or USP36-deficient hFOB1.19 cells were seeded into 96-well plates at a density of 8 × 103 cells/well, and cultured for 0, 24, 48, or 72 h. Then, the cells were incubated with 0.5% MTT for 3 hours and treated with 100 µL DMSO on a shaker for 15 min. Finally, the optical density (OD) values at 490 nm were measured by a microplate reader.
Cell cycle detection
7-aminoactinomycin D (7-AAD) staining assay was carried out for cell cycle detection. In brief, control or USP36-deficient hFOB1.19 cells were seeded into 6-well plates at a density of 3 × 105 cells/well and cultured for 24 h, followed by fixation using ice-cold 70% ethanol for 6 h at 4 °C. Subsequently, the fixed cells were washed twice with PBS and treated with PBS containing 0.2% Triton X-100 and 10 µg/ml RNase at 37 °C for 30 min. After that, the cells were stained with 20 mg/mL 7-AAD solution (Beyotime Biotechnology, Haimen, China) at RT for 30 min in the dark. Finally, the cells were subject to flow cytometry using a NovoCyte flow cytometer (Agilent Technologies, Santa Clara, CA, USA), and the data were analyzed by NovoExpress® software 1.4.1 (Agilent Technologies).
qPCR
Total RNA was extracted from hFOB1.19 cells using the TRIzol reagent (Invitrogen) and was reverse transcribed into cDNA with a HiScript II 1st Strand cDNA Synthesis kit (Vazyme, Nanjing, China), according to the manufacturer’s instructions. qPCR was carried out using a ChamQ SYBR qPCR Master Mix kit (Vazyme) on an iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). The expression of target genes was normalized with 18S RNA using the 2−ΔΔCT method. The primer sequences for qPCR are listed in Table 2.
Apoptosis detection
Annexin V/PI staining was employed to assess the apoptosis of control and USP36-deficient cells using the Annexin V FITC Kit (Beyotime Biotechnology) following the manufacturer’s guide. In brief, 1 × 106 cells were seeded into each well of 6-well plates. After 24 h of culture, the cells were collected and treated with the Annexin V-FITC/PI working solution prepared from the kit in the dark for 15 min. Finally, the cells were analyzed using a NovoCyte flow cytometer (Agilent Technologies), and the results were analyzed with NovoExpress® software 1.4.1 (Agilent Technologies).
Western blotting
Cells were lysed with RIPA buffer containing phosphatase inhibitors (Beyotime Biotechnology), with homogenization on ice. After centrifugation, the supernatants were collected, and the protein concentration was determined using a BCA kit (PA115-02; TIANGEN Biotechnology, Beijing, China). Then the samples were denatured, separated via 10% SDS-PAGE, and transferred onto Polyvinylidene fluoride (PVDF, Sigma-Aldrich) membranes. Subsequently, the membranes were blocked with 5% skimmed milk at RT for 1 h, followed by 2 h of incubation with primary antibody solutions at RT. Next, the membranes were washed with 1X Tris-Buffered Saline + 0.1% Tween 20 (TBST, Sigma-Aldrich) three times and incubated with the secondary antibody solution for 1 h at RT. After three washes of TBST, the membranes were incubated with an enhanced chemiluminescent kit (Thermo Fisher Scientific, Waltham, MA, USA), and the signal was visualized and documented by a ChemiDoc imaging system (Bio-Rad Laboratories). The band intensity was quantified by ImageJ v1.48 (NIH, Bethesda, MD, USA). The antibody information is present in Table 3.
Co-immunoprecipitation (Co-IP)
The coding sequence of USP36 and WDR5 was inserted into pcDNA3.1-HA or pcDNA3.1-Flag Plasmids, respectively, and named HA-USP36 and Flag-WDR5 (WDR5 OE). They were co-transfected into hFOB1.19 cells to elucidate the interaction between USP36 and WDR5. To understand the impact of USP36 on WDR5 ubiquitination, shUSP36 and/or Flag-WDR5 expression plasmids were co-transfected with His-tag conjugated ubiquitin (His-Ub) plasmid into hFOB1.19 cells. HA-USP36, Flag-WDR5, and His-Ub plasmids were purchased from Miaoling Biotechnology (Wuhan, China), and were sequenced to confirm the accuracy of the DNA sequence. 24 h after transfection, hFOB1.19 cells were collected, and cell lysates were prepared and subjected to co-IP experiments according to the instructions of the Flag-tag Protein IP Assay Kit with Magnetic Beads (Beyotime Biotechnology) and the HA-tag Protein IP Assay Kit with Magnetic Beads (Beyotime Biotechnology). The antibody information is listed in Table 3.
Statistical analysis
All experiments were performed in triplicates in this study for quantification purposes unless otherwise indicated. Bar chart generation and statistical analysis were done by Prism (version 8.0, GraphPad Software, Boston, MA, USA). Data are presented as means with standard deviations. Shapiro–Wilk test was employed to evaluate the normality of the data. Two-tailed Student’s t-test (unpaired) was used to compare the difference between two groups of data, while One-way ANOVA followed by Tukey’s post-hoc test was used to identify the significant differences among multiple groups of data. P < 0.05 is considered significant.
Results
USP36 is highly expressed during hFOB1.19 cell differentiation
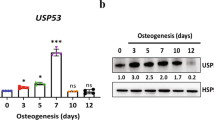
To screen USPs that may regulate osteoblast differentiation, we initially evaluated the expression of 48 USPs in hFOB1.19 cells, which are capable of undergoing osteogenic differentiation upon treatment with osteogenic medium [19, 20]. Alizarin red staining confirmed successful osteogenic differentiation in hFOB1.19 cells after 21 days of culture (Fig. 1A). Subsequently, we compared the expression of these 48 USPs between differentiated and undifferentiated hFOB1.19 cells using qPCR, revealing significant changes in the expression of multiple USPs during osteogenic differentiation. Notably, USP36 exhibited the most substantial increase in expression (Fig. 1B), suggesting its potential role in regulating osteogenic differentiation in hFOB1.19 cells.

USP36 expression is strongly induced in hFOB1.19 cells undergoing osteogenic differentiation. (A) The morphology of undifferentiated hFOB1.19 cells and Alizarin red-stained hFOB1.19 cells cultured with the osteogenic medium for 21 days. Note the presence of mineralized nodules (yellow arrowheads). (B) The qPCR results indicated the expression ratio of each USP in differentiated hFOB1.19 cells compared to undifferentiated hFOB1.19 cells
USP36 deficiency caused reduced proliferation, cell cycle arrest, and increased apoptosis of hFOB1.19 cells
To investigate the role of USP36 in hFOB1.19 cells, we constructed three shUSP36 variants and observed that all of them efficiently reduced USP36 mRNA levels in transfected hFOB1.19 cells when compared to shNC (Fig. 2A). Given that shUSP36-1 displayed the highest knockdown efficiency, we selected it to generate USP36-deficient hFOB1.19 cells. We observed a significant downregulation in the proliferation of hFOB1.19 cells upon USP36 knockdown, as indicated by the results of the MTT assay (Fig. 2B). Furthermore, USP36 deficiency resulted in cell cycle arrest, as evidenced by 7-AAD staining results (Fig. 2C and D). Additionally, Annexin V/PI staining data revealed that the knockdown of USP36 significantly increased the apoptosis of hFOB1.19 cells (Fig. 2E and F). These findings suggest that USP36 plays a critical role in controlling the proliferation, cell cycle, and survival of hFOB1.19 cells.

USP36 knockdown causes decreased proliferation, cell cycle arrest, and increased apoptosis in hFOB1.19 cells. (A) qPCR data showed that all three shUSP36s could efficiently reduce the mRNA level of USP36 in hFOB1.19 cells. (B) MTT assay results indicated that USP36-deficient hFOB1.19 cells were less proliferative than control hFOB1.19 cells. (C) 7-AAD staining data showed that USP36 knockdown resulted in cell cycle arrest in hFOB1.19 cells. (D) Quantification of the 7-AAD staining data present in (C). (E) Annexin V/PI staining results suggested that the apoptosis of hFOB1.19 cells was increased upon USP36 deficiency. (F) Quantification of the results in (E). NC, negative control
USP36 knockdown impaired the osteogenic differentiation of hFOB1.19 cells
To determine whether USP36 regulates the osteogenic differentiation of hFOB1.19 cells, we cultured both control and USP36-deficient hFOB1.19 cells in the presence of osteogenic medium. Our observations revealed that USP36-deficient cells formed fewer calcified nodules than control cells, as demonstrated by Alizarin red staining (Fig. 3A and B). Furthermore, we noted a significant reduction in the levels of OCN, RUNX2, Collagen I, and ALP, which serve as markers for osteogenic differentiation [21, 22], in USP36-deficient cells compared to control cells, as evidenced by qPCR and Western blotting data (Fig. 3C and E). These results conclusively establish the pivotal role of USP36 in the osteogenic differentiation of hFOB1.19 cells.

USP36 deficiency disrupts the osteogenic differentiation of hFOB1.19 cells. (A) Alizarin red staining data showed that USP36-deficient hFOB1.19 cells formed fewer nodules than control cells. (B) Quantification of the results in (A). (C) qPCR results indicated that the mRNA levels of USP36, OCN, RUNX2, Collagen I, and ALP were significantly downregulated in hFOB1.19 cells cultured with the osteogenic medium for 21 days upon USP36 deficiency. (D) Western blotting data uncovered that the protein levels of USP36, OCN, RUNX2, Collagen I, and ALP were also remarkably reduced in hFOB1.19 cells cultured with the osteogenic medium for 21 days upon USP36 knockdown. (E) Quantification of the Western blotting data shown in (D). NC, negative control
USP36 interacts with WDR5 and regulates its ubiquitination
To elucidate the mechanism underlying the regulatory effect of USP36 on the osteogenic differentiation of hFOB1.19 cells, we searched the BioGRID database to screen its potential substrates [23]. We selected WD Repeat-Containing Protein 5 (WDR5) as it plays an essential role in regulating osteoblast differentiation [24, 25]. Indeed, co-IP experiment results revealed an interaction between USP36 and WDR5 in hFOB1.19 cells (Fig. 4A). To determine whether USP36 influences the ubiquitination of WDR5, we knocked down USP36 in hFOB1.19 cells and concurrently introduced His-Ub. IP data indicated that USP36 deficiency caused an increased level of WDR5 (Fig. 4B). To further explore whether USP36 controls the stability of WDR5 protein, we treated control and USP36-deficient hFOB1.19 cells with protein synthesis inhibitor cycloheximide (CHX). Because CHX blocks WDR5 synthesis, we can measure the stability of existing WDR5. Western blotting results showed that USP36 knockdown significantly accelerated the degradation of WDR5 in hFOB1.19 cells (Fig. 4C). These results demonstrate that USP36 could interact with and stabilize WDR5 by promoting its deubiquitination level.

USP36 interacts with WDR5 protein and regulates its ubiquitination and stability in hFOB1.19 cells. (A) Co-IP assay results showed an interaction between USP36 and WDR5. (B) IP assay data revealed that knockdown of USP36 resulted in an increased level of WDR5 ubiquitination. (C) Western blotting assays indicated that knockdown of USP36 promoted the degradation of the Flag-WDR5 protein. TCL, total cell lysate
Excessive WDR5 rescues the mitigated osteogenic differentiation of USP36-deficient hFOB1.19 cells
To determine whether USP36 controls the osteogenic differentiation of hFOB1.19 cells by regulating the expression of WDR5, we overexpressed WDR5 in USP36-deficient hFOB1.19 cells and observed their osteogenic differentiation by Alizarin red staining. The results showed that overexpression of WDR5 significantly increased the number of mineralized nodules formed by USP36-deficient cells to a level comparable to that formed by control cells (shNC) (Fig. 5A and B). These results further support the notion that USP36 regulates the osteogenic differentiation of hFOB1.19 cells by stabilizing WDR5.

Overexpression of WDR5 rescued the osteogenic differentiation of USP36-deficient hFOB1.19 cells. (A) Alizarin red staining data showed that USP36 knockdown hFOB1.19 cells formed fewer mineralized nodules compared to control cells, while overexpression of WDR5 rescued this defect. (B) Quantification of the results in (A)
Discussion
More than 50 USPs have been identified in the human genome, and they regulate various physiological and pathological processes [26, 27]. As previously mentioned, multiple USPs have been implicated in regulating the function and osteogenic differentiation of osteoblasts. Given the large number of USPs, we speculate that other USPs may also contribute to the regulation of osteoblasts. Therefore, we evaluated the expression of 48 USPs in differentiated and undifferentiated hFOB1.19 osteoblasts.
Notably, we found that the expression of USP4, which represses osteoblast differentiation, was significantly decreased in differentiated hFOB1.19 cells, consistent with its inhibitory role in osteoblast differentiation. Interestingly, USP15 expression was also markedly downregulated in differentiated hFOB1.19 cells, possibly because the cells no longer require the pro-differentiation function of USP15. Meanwhile, the expression of USP7 was also remarkably reduced in hFOB1.19 cells that underwent osteogenic differentiation.
On the other hand, we identified multiple USPs that were greatly overexpressed in differentiated hFOB1.19 cells. In particular, USP36 was the most upregulated USP, and its function in regulating osteoblast activities remains unclear. A similar increase in USP36 expression was observed in MC3T3-E1 osteoblasts undergoing osteogenic differentiation triggered by intermittent treatment with PTH [28]. These results indicate that USP36 may function as a vital regulator of osteoblast function and activities. Indeed, our functional analyses of USP36-deficient hFOB1.19 cells provide direct evidence that USP36 promotes the proliferation, survival, and differentiation of hFOB1.19 cells.
USP36 was initially isolated and characterized from ovarian cancer cells [29], and it can stabilize ALKBH5, c-MYC, SOD2, and YAP to control tumorigenesis, drug sensitivity, and mitochondrial oxidative stress [30,31,32,33]. Knockdown of USP36 represses proliferation and induces apoptosis in multiple cell types, not limited to osteoblast [34], consistent with its role in osteoblast. These results suggest that USP36 may regulate cell proliferation and survival through a common mechanism across different cell types.
To elucidate the mechanism by which USP36 regulates osteoblast activities, we investigated its impact on WDR5. WDR5 accelerates osteoblast differentiation by directly binding to the promoter of Wnt1 and its target genes c-Myc and Runx2, thereby facilitating their expression [25]. However, the mechanism controlling WDR5 itself, particularly at the post-translational level in osteoblasts, remains incompletely understood. It has been reported that WDR5 ubiquitination is promoted by the SKP1-CUL1-F-Box E3 ubiquitin ligase complex and E3 ligase RNF220 in different contexts [35, 36]. By comparison, deubiquitinases like USP44 remove WDR5 ubiquitination in other scenarios [37]. Our finding that USP36 decreases the ubiquitination of WDR5 supports the notion that WDR5 stability is tightly controlled at the post-translational level. However, the interacting domains of USP36 and WDR5, and the type of ubiquitination (such as K48-linked and/or K63-linked ubiquitination) of WDR5 controlled by USP36, require further characterization.
Interestingly, WDR5 can activate c-MYC transcription, as mentioned above, and USP36 may also directly deubiquitinate c-MYC in hFOB1.19 cells, similar to its action in another context [32]. This suggests a vital role of c-MYC in mediating the influence of the USP36-WDR5 axis on osteoblast activities. Nevertheless, given the diverse substrates of USP36, it is possible that USP36 regulates osteoblast activities by regulating other targets. Further investigations are warranted to explore these possibilities.
In summary, our data reveal that USP36 is a potent regulator of osteoblast function and activity and that the mechanism of its regulatory action on osteoblast function is closely related to the reduction of ubiquitination levels in WDR5. These findings expand our knowledge about the function of USPs in osteoblast biology. Nevertheless, our conclusion is solely based on investigation in hFOB1.19 cells, it will be essential to verify these findings in other osteoblast cell lines and mouse models.
Data availability
The data supporting the conclusions of this article have been included in the manuscript.
References
Henry JP, Bordoni B, Histology. Osteoblasts. StatPearls. Treasure Island (FL)2023.
Marie PJ. Osteoblast dysfunctions in bone diseases: from cellular and molecular mechanisms to therapeutic strategies. Cell Mol Life Sci. 2015;72(7):1347–61. https://doi.org/10.1007/s00018-014-1801-2. Epub 20141209.
Rutkovskiy A, Stenslokken KO, Vaage IJ. Osteoblast differentiation at a glance. Med Sci Monit Basic Res. 2016;22:95–106. Epub 20160926. doi: 10.12659/msmbr.901142. PubMed PMID: 27667570; PubMed Central PMCID: PMCPMC5040224.
Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280(39):33132–40. https://doi.org/10.1074/jbc.M500608200. Epub 20050725.
Phimphilai M, Zhao Z, Boules H, Roca H, Franceschi RT. BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J Bone Min Res. 2006;21(4):637–46. https://doi.org/10.1359/jbmr.060109. Epub 20060405.
Shimoyama A, Wada M, Ikeda F, Hata K, Matsubara T, Nifuji A, et al. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol Biol Cell. 2007;18(7):2411–8. https://doi.org/10.1091/mbc.e06-08-0743. Epub 20070418.
Wang BL, Dai CL, Quan JX, Zhu ZF, Zheng F, Zhang HX, et al. Parathyroid hormone regulates osterix and Runx2 mRNA expression predominantly through protein kinase A signaling in osteoblast-like cells. J Endocrinol Invest. 2006;29(2):101–8. https://doi.org/10.1007/BF03344081. PubMed PMID: 16610234.
Battaglino R, Kim D, Fu J, Vaage B, Fu XY, Stashenko P. c-myc is required for osteoclast differentiation. J Bone Min Res. 2002;17(5):763–73. .763. PubMed PMID: 12009006.
Zhang C, Cho K, Huang Y, Lyons JP, Zhou X, Sinha K, et al. Inhibition of wnt signaling by the osteoblast-specific transcription factor Osterix. Proc Natl Acad Sci U S A. 2008;105(19):6936–41. https://doi.org/10.1073/pnas.0710831105. Epub 20080505.
Mulder MP, Witting K, Berlin I, Pruneda JN, Wu KP, Chang JG, et al. A cascading activity-based probe sequentially targets E1-E2-E3 ubiquitin enzymes. Nat Chem Biol. 2016;12(7):523–30. https://doi.org/10.1038/nchembio.2084. Epub 2016/05/18.
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–86. PubMed PMID: 16325574.
Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373(6509):81–3. https://doi.org/10.1038/373081a0. Epub 1995/01/05.
Zhou F, Li F, Fang P, Dai T, Yang B, van Dam H, et al. Ubiquitin-specific protease 4 antagonizes osteoblast differentiation through dishevelled. J Bone Min Res. 2016;31(10):1888–98. https://doi.org/10.1002/jbmr.2863. Epub 20160520.
Greenblatt MB, Shin DY, Oh H, Lee KY, Zhai B, Gygi SP, et al. MEKK2 mediates an alternative beta-catenin pathway that promotes bone formation. Proc Natl Acad Sci U S A. 2016;113(9):E1226–35. https://doi.org/10.1073/pnas.1600813113. Epub 20160216.
Guo YC, Wang MY, Zhang SW, Wu YS, Zhou CC, Zheng RX et al. Ubiquitin-specific protease USP34 controls osteogenic differentiation and bone formation by regulating BMP2 signaling. EMBO J. 2018;37(20). Epub 20180904. https://doi.org/10.15252/embj.201899398. PubMed PMID: 30181118; PubMed Central PMCID: PMCPMC6187217.
Kim JM, Yang YS, Park KH, Ge X, Xu R, Li N, et al. A RUNX2 stabilization pathway mediates physiologic and pathologic bone formation. Nat Commun. 2020;11(1):2289. https://doi.org/10.1038/s41467-020-16038-6. Epub 20200508.
Ji L, Lu B, Zamponi R, Charlat O, Aversa R, Yang Z, et al. USP7 inhibits Wnt/beta-catenin signaling through promoting stabilization of Axin. Nat Commun. 2019;10(1):4184. https://doi.org/10.1038/s41467-019-12143-3. Epub 20190913.
Li J, Hu L, Liu Y, Huang L, Mu Y, Cai X et al. DDX19A Senses Viral RNA and Mediates NLRP3-Dependent Inflammasome Activation. Journal of immunology (Baltimore, Md: 1950). 2015;195(12):5732-49. Epub 2015/11/06. https://doi.org/10.4049/jimmunol.1501606. PubMed PMID: 26538395.
Mizerska-Kowalska M, Slawinska-Brych A, Kalawaj K, Zurek A, Pawinska B, Rzeski W et al. Betulin Promotes Differentiation of Human Osteoblasts In Vitro and Exerts an Osteoinductive Effect on the hFOB 1.19 Cell Line Through Activation of JNK, ERK1/2, and mTOR Kinases. Molecules. 2019;24(14). Epub 20190719. https://doi.org/10.3390/molecules24142637. PubMed PMID: 31331121; PubMed Central PMCID: PMCPMC6680433.
Harris SA, Enger RJ, Riggs BL, Spelsberg TC. Development and characterization of a conditionally immortalized human fetal osteoblastic cell line. J Bone Min Res. 1995;10(2):178–86. .5650100203. PubMed PMID: 7754797.
Komori T. Regulation of proliferation, differentiation and functions of osteoblasts by Runx2. Int J Mol Sci. 2019;20(7):1694. https://doi.org/10.3390/ijms20071694. Epub 2019/04/17.
Karsenty G. Minireview: transcriptional control of osteoblast differentiation. Endocrinology. 2001;142(7):2731–3. https://doi.org/10.1210/endo.142.7.8306. Epub 2001/06/21.
Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021;30(1):187–200. Epub 20201123. doi: 10.1002/pro.3978. PubMed PMID: 33070389; PubMed Central PMCID: PMCPMC7737760.
Gori F, Friedman LG, Demay MB. Wdr5, a WD-40 protein, regulates osteoblast differentiation during embryonic bone development. Dev Biol. 2006;295(2):498–506. https://doi.org/10.1016/j.ydbio.2006.02. Epub 20060530.
Zhu ED, Demay MB, Gori F. Wdr5 is essential for osteoblast differentiation. J Biol Chem. 2008;283(12):7361-7. Epub 20080116. https://doi.org/10.1074/jbc.M703304200. PubMed PMID: 18201971.
Young MJ, Hsu KC, Lin TE, Chang WC, Hung JJ. The role of ubiquitin-specific peptidases in cancer progression. J Biomed Sci. 2019;26(1):42. https://doi.org/10.1186/s12929-019-0522-0. Epub 20190527.
Li Y, Reverter D. Molecular mechanisms of DUBs Regulation in Signaling and Disease. Int J Mol Sci. 2021;22(3). Epub 20210120. doi: 10.3390/ijms22030986. PubMed PMID: 33498168; PubMed Central PMCID: PMCPMC7863924.
Hariri H, St-Arnaud R. Expression and role of ubiquitin-specific peptidases in Osteoblasts. Int J Mol Sci. 2021;22(14). Epub 20210720. doi: 10.3390/ijms22147746. PubMed PMID: 34299363; PubMed Central PMCID: PMCPMC8304380.
Kim MS, Kim YK, Kim YS, Seong M, Choi JK, Baek KH. Deubiquitinating enzyme USP36 contains the PEST motif and is polyubiquitinated. Biochem Biophys Res Commun. 2005;330(3):797–804. https://doi.org/10.1016/j.bbrc.2005.03.051. Epub 2005/04/06.
Chang G, Xie GS, Ma L, Li P, Li L, Richard HT. USP36 promotes tumorigenesis and drug sensitivity of glioblastoma by deubiquitinating and stabilizing ALKBH5. Neurooncology. 2023;25(5):841–53. https://doi.org/10.1093/neuonc/noac238. Epub 2022/10/15.
Kim MS, Ramakrishna S, Lim KH, Kim JH, Baek KH. Protein stability of mitochondrial superoxide dismutase SOD2 is regulated by USP36. J Cell Biochem. 2011;112(2):498–508. https://doi.org/10.1002/jcb.22940. Epub 2011/01/27.
Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc Natl Acad Sci U S A. 2015;112(12):3734–9. https://doi.org/10.1073/pnas.1411713112. Epub 2015/03/17.
Zhang W, Luo J, Xiao Z, Zang Y, Li X, Zhou Y, et al. USP36 facilitates esophageal squamous carcinoma progression via stabilizing YAP. Cell Death Dis. 2022;13(12):1021. https://doi.org/10.1038/s41419-022-05474-5. Epub 2022/12/06.
Fraile JM, Campos-Iglesias D, Rodríguez F, Astudillo A, Vilarrasa-Blasi R, Verdaguer-Dot N, et al. Loss of the deubiquitinase USP36 destabilizes the RNA helicase DHX33 and causes preimplantation lethality in mice. J Biol Chem. 2018;293(6):2183–94. https://doi.org/10.1074/jbc.M117.788430. Epub 2017/12/24.
Hanle-Kreidler S, Richter KT, Hoffmann I. The SCF-FBXW7 E3 ubiquitin ligase triggers degradation of histone 3 lysine 4 methyltransferase complex component WDR5 to prevent mitotic slippage. J Biol Chem. 2022;298(12):102703. https://doi.org/10.1016/j.jbc.2022.102703. Epub 20221114.
Wang H, Liu X, Liu Y, Yang C, Ye Y, Sheng N, et al. The E3 ubiquitin ligase RNF220 maintains hindbrain < i > hox expression patterns through regulation of WDR5 stability. Cold Spring Harbor Laboratory; 2023.
Chi Z, Zhang B, Sun R, Wang Y, Zhang L, Xu G. USP44 accelerates the growth of T-cell acute lymphoblastic leukemia through interacting with WDR5 and repressing its ubiquitination. Int J Med Sci. 2022;19(14):2022–32. https://doi.org/10.7150/ijms.74535. Epub 20221114.
Acknowledgements
N/A.
Funding
This study was supported by Xiamen Medical and Health Guidance Projects (No. 3502Z20214ZD1094).
Author information
Authors and Affiliations
Contributions
J.F.Y: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Roles/Writing - original draft, Writing - review & editing. X.F.G: Investigation, Methodology, Validation, Visualization, Writing - review & editing. X.L.G: Conceptualization, Investigation, Methodology, Validation, Writing - review & editing. Y.S: Data curation, Investigation, Methodology, Validation, Writing - review & editing. M.H.J: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing - review & editing.
Corresponding author
Ethics declarations
Ethics approval/consent to participate
N/A.
Consent for publication
N/A.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, J., Gu, X., Gao, X. et al. USP36 regulates the proliferation, survival, and differentiation of hFOB1.19 osteoblast. J Orthop Surg Res 19, 483 (2024). https://doi.org/10.1186/s13018-024-04893-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-024-04893-8