
Abstract
The diagnostic odysseys for rare disease patients are getting shorter as next-generation sequencing becomes more widespread. However, the complex genetic diversity and factors influencing expressivity continue to challenge accurate diagnosis, leaving more than 50% of genetic variants categorized as variants of uncertain significance.
Genomic expression intricately hinges on localized interactions among its products. Conventional variant prioritization, biased towards known disease genes and the structure-function paradigm, overlooks the potential impact of variants shaping the composition, location, size, and properties of biomolecular condensates, genuine membraneless organelles swiftly sensing and responding to environmental changes, and modulating expressivity.
To address this complexity, we propose to focus on the nexus of genetic variants within biomolecular condensates determinants. Scrutinizing variant effects in these membraneless organelles could refine prioritization, enhance diagnostics, and unveil the molecular underpinnings of rare diseases. Integrating comprehensive genome sequencing, transcriptomics, and computational models can unravel variant pathogenicity and disease mechanisms, enabling precision medicine. This paper presents the rationale driving our proposal and describes a protocol to implement this approach. By fusing state-of-the-art knowledge and methodologies into the clinical practice, we aim to redefine rare diseases diagnosis, leveraging the power of scientific advancement for more informed medical decisions.
Similar content being viewed by others


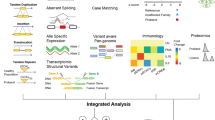
The idea
The diagnosis of rare diseases (RDs) remains a challenging and complex endeavor. The genetic diversity present among the nearly 8 billion living humans, with 5·106 variants on average [1], hinders the understanding of genetic traits [2, 3]. This is reinforced by the intricate genetic regulation and the complex interplay of factors that modulate expressivity in RDs. Therefore, it is unsurprising that more than half of genetic variants are considered variants of uncertain significance (VUS) [4, 5], with patients of non-European descendants bearing the brunt [6, 7]. In RDs, this abundance of VUSs is especially significant because it is estimated that around 80% of them have a genetic basis [8].
In the last two decades, genetics has made significant progress, revealing new gene-disease associations, causative molecular mechanisms, and therapeutic developments [9]. However, the intricate interplay of extensive genetic diversity, variable expressivity, and incomplete penetrance in RDs hampers genetic diagnosis and the establishment of clinical relevance in variants related to conditions. A task that is further complicated by the influence of genetic background diversity, epigenetic modifications, environmental factors, and the limited number of cases [10]. Even identical causal variants over different genetic backgrounds can lead to diverse pathological phenotypes, This complexity requires physicians to apply “clinical diagnostic criteria” a set of phenotypic characteristics that patients with a given genetic variant may exhibit in different numbers and degrees. For example, a patient with a pathogenic variant in PTPN11 (MIM #176876) causing Noonan syndrome may have pulmonary stenosis, while a different patient with the same variant may have a different congenital heart disease, or even no heart disease but shows many other Noonan syndrome characteristics [11]. Conversely, some individuals with disease-causing alleles remain healthy despite affected family members in the same environment [10, 12]. Moreover, the lack of comprehensive studies and adequate tools to gather and analyze this information hinders our ability to fully understand the pathological significance of genetic variants. As a result, diagnosing RDs remains a relevant challenge despite advances in genetic medicine [13].
Identifying the causative variant and mode of inheritance is mandatory to guide a patient’s clinical management, inform patients about related risks, and aid in evaluating family planning options. Unfortunately, diagnosis is long delayed, depending on the patient’s phenotype, age, and resources. On average, it takes around 4–5 years to accurately diagnose a specific RD, but in some cases, a definitive diagnosis can take more than a decade or even die without it [14,15,16,17]. Patients often undergo costly and extensive evaluations at multiple institutions and may remain undiagnosed or misdiagnosed, causing emotional distress to patients and relatives. Fortunately, as our understanding of mechanisms behind phenotypic causation advances, new pieces of the puzzle emerge, and we will become better equipped to generate and experimentally verify hypotheses regarding the origins of this phenotypic variability.
The reductionist approach in prioritizing variants, focusing only on well-known disease-causing genes, hinders genetic diagnosis. Despite Fisher’s seminal work proposing polygenic inheritance in 1918 [18] and later validated [19, 20], most variant prioritization algorithms persist in a gene-to-gene approach. But, navigating the complex pathways connecting genotypes to phenotypes requires more comprehensive approaches to avoid uncertain significance scenarios. Genome-wide association studies have further supported the necessity of a systemic view by demonstrating that common SNPs contribute to the genetic architecture of multifactorial traits [21,22,23]. These variants may affect genes not directly linked to a specific disease, but their cumulative effect may ultimately impact the resulting phenotype [24]. Our adherence could be due to our limited comprehension of the emergent properties arising from epistatic interactions, as well as the need to facilitate clinical management.
Although we are starting to analyze epistatic cross-regulation mediated by nonadditive gene-to-gene interactions, this remains largely unexplored in the prioritization of genetic variants. The individual actions of each of our genes are limited, but collective behavior arises as a result of their local interactions, giving rise to a complex organization [25,26,27]. Thereby, we must consider the genome as a whole, without overlooking that it comprises individual pieces that coordinate this collective behavior. This complexity may hinder the precise elucidation of the exact number of genes involved and their contributions to phenotypes. To comprehensively understand complex traits, alternative approaches beyond current genotypic analysis are needed. Therefore, we propose an open-minded approach, exploring innovative strategies and thoroughly investigating all variants impacting specific molecular self-organization.
Analyzing patient variants requires considering gene products beyond transcriptional regulation or catalytic activities. Proteins, RNAs, or their combinations operate in crowded environments with competitive molecular interactions. Understanding the collective behavior of genetic diseases relies on two key elements: intrinsically disordered regions (IDRs) [28] and biomolecular condensates (BCs) [29]. Both IDRs and BCs have emerged as significant contenders in unraveling the mysteries of conditions such as cancer, neurodegenerative disorders, or RDs [30,31,32,33,34,35]. The dysregulation of IDRs and BCs presents an intriguing enigma (see Fig. 1) that requires further exploration. However, their integration into routine clinical practice remains unexplored despite their potential impact. Beyond diagnostic and therapeutic applications, IDRs and BCs offer valuable insights into complex phenomena like variable expressivity and epistasis, which are characteristic of RDs.
Why explore the effects of variants in IDRs?
In the 1960s, the experiments of biochemist Christian B. Anfinsen established the sequence-structure-function paradigm [36]. The sequence-structure-function paradigm proposes that a protein’s primary amino acid sequence dictates its three-dimensional structure and function. However, biology has shown exceptions, such as the IDRs or intrinsically disordered proteins (IDPs) that lack a fixed three-dimensional structure and exhibit a wide range of conformations and functions [28, 37].
Unlike the conventional protein structure-function model, half of the proteome still performs cellular functions without fully or partially well-defined three-dimensional structures under physiological conditions [38, 39]. In humans, fully folded proteins (37%) or IDPs (5%) only represent the two ends of the continuum [40]. Most human proteins (58%) contain folded protein domains and IDRs [41]. But, so far, little attention has been paid to IDRs or IDPs [37]. However, IDRs deviate from the classical paradigm and have led to the emergence of the disorder-function paradigm that postulates that proteins can remain unfolded while still carrying out their essential physiological functions [34].
IDRs exhibit a captivating functional diversity, spanning up to 8 distinct subtypes [42]. Of particular interest is the fact that these functional subtypes may individually manifest or coexist within a single protein, showing the intricate complexity that can arise from the convergence of multiple disordered regions. This interplay among IDRs highlights their pivotal role in shaping the multifaceted functionality of proteins.
Additional evidence suggests that we should pay more attention to IDRs. Computational analyses estimate that approximately 14% of the proteome in archaea and bacteria, and a substantial portion ranging from 44 to 54% in eukaryotes, consists of disordered regions [43]. Moreover, evolutionary trends reveal that as the genome’s complexity increases, so does the proportion of IDRs within the proteome, particularly during the transition from prokaryotic to eukaryotic life [44, 45]. Furthermore, IDRs tend to be enriched in proteins performing complex functions like signaling, while they are depleted in proteins with more structure-dependent functions, such as metabolic proteins [39, 46]. The fundamental attribute of these sequences is their capacity to regulate and modify protein activity, enabling adaptive responses to diverse situations. This is achieved thanks to the conformational heterogeneity of facilitating proteins, which influences their interactions with other molecules [34].
Notably, IDRs are estimated to be involved in over 20% of genetic diseases on average but can be increased to 50%, such as in skeletal disorders [40]. Focusing on disordered charged biased proteins, 95% of them are associated with multiple diseases [47]. Furthermore, up to 25% of documented disease mutations have been identified within IDRs [48]. Mutations in IDPs such as β-amyloid, α-synuclein, and FUS, have emerged as key contributors to a spectrum of neurodegenerative diseases [49]. These alterations disrupt the interaction dynamics of these IDPs, prompting their aberrant aggregation and, consequently, instigating the pathogenesis of disorders such as Alzheimer’s Disease [49], Parkinson’s Disease [50], or amyotrophic lateral sclerosis [51].
Moreover, the adaptive and regulatory capacity of IDRs is further supported by observed facts related to alternative splicing events [34, 52,53,54]. Proteins containing tissue-specific exons exhibit a higher average number of interaction partners and serve as central hubs in protein-protein interaction networks [34, 53]. Furthermore, these differentially expressed exons are enriched in IDRs [53]. In addition, IDRs exhibit conserved linear interactive motifs and post-translational modification sites. Hence, tissue-specific splicing of exons facilitates the rewiring of protein-protein interaction networks, enabling adaptation to environmental cues and changing the nature of the response itself. IDRs evolve faster than structured segments due to the reduced constraints on amino acid substitution [55,56,57,58], resulting in a higher frequency of variants in these regions.
Structured domains and IDRs should be considered as two functional components of proteins [42]. However, variants within IDRs are often underestimated, leading to their frequent classification as VUS, as a result of the structure-function paradigm use. This limitation hinders our understanding of protein functionality and its implications for human health. The up to now presented facts leave unanswered questions about the functional consequences of variants impacting IDRs in disease contexts and how those IDRs execute specific functions without well-defined structures. Variant-interpretation criteria are applied regardless of whether the region is structured or disordered. Studies carried out in the field of cancer and evolution, point out that folded domains and IDRs differ in terms of their tolerance to mutations [58]. IDRs can display higher tolerance to sequence variations, as they don’t rely on a specific structure to function. However, residues involved in interactions or post-translational modification (PTM) sites within IDRs exhibit similar constraints as globular proteins [58].
Despite the extensive evidence, studies tend to focus on mutations within folded regions, sometimes neglecting or classifying mutations within IDRs as VUS. Considering these findings, it is crucial to thoroughly study the effects of variants in IDRs when prioritizing variants of RDs.

Schematic representation of the factors that modulate the LLPS dynamics in the formation of biomolecular condensates and the potential consequences of variants affecting IDRs. LCRs: The low-complexity regions exhibit a limited range of amino acid compositions, leading to reduced amino acid diversity within these regions. Locally, amino acids tend to cluster, forming hydrophobic or electrostatic patches that facilitate the molecular aggregation process; SLiMs: short linear interacting motives, PTMs: Post-translational modifications, MID: modular interacting domain, IDR: Intrinsically disordered regions, CC: critical concentration
Why explore the effect of variants in BCs?
Within the crowded cellular milieu, processes require precise spatiotemporal regulation and organization. Conventionally, this organization has been attributed to lipid membrane organelles. However, the emerging concept of biomolecular condensation demonstrates that fundamental cellular biochemistry extends beyond membrane barriers [29, 59]. These BCs selectively concentrate biomolecules in defined foci, leading to membraneless organelles (MLOs). The absence of membranes in these condensates facilitates rapid sensing and adaptation to environmental changes, allowing the exchange of their constituents with the surrounding cytoplasm or nucleoplasm without requiring specialized transporters [51]. The emergence of MLOs is mainly attributed to the liquid-liquid phase separation (LLPS) in biomolecules [60, 61] (see Fig. 1). Through this organizational mechanism, cells create unique environments by selecting specific components that regulate biomolecule availability, reducing noise in cellular computation and facilitating enhancing reaction rates [62,63,64]. This dynamic regionalization of components enables the precise orchestration of a myriad of cellular reactions and processes. BCs have thus emerged as primary organizers at different scales, and their role in both physiological and pathological processes has been fully demonstrated [32, 33, 60, 65,66,67].
BCs acting as hubs for signal modulation add another layer of complexity to phenotype determination [68, 69]. Therefore, understanding the interplay between variable expressivity and BCs is vital for deciphering pathological mechanisms and phenotypic heterogeneity in RDs. However, to the best of our knowledge, variant prioritization algorithms that address this have not yet been developed for RDs. This pending task can be facilitated by filtering variants as affecting scaffolds or clients [70, 71]. Scaffolds are biomolecules that self-associate through multivalent interactions, driving molecular condensation, while clients join this scaffolding, modulating the condensate’s composition and creating a liquid network of competitive interactions [72]. This process causes interacting components to segregate, leading to a condensed phase with higher biomolecule density, analogous to precipitation in saturated solutions. Specific proteins exhibit fundamental characteristics that promote condensate formation [51, 73,74,75,76].
IDPs and IDRs play a crucial role in cellular homeostasis through molecular condensation processes [29, 77]. In each protein, the primary structures and particular portions of linear sequences within it, as low complexity regions (LCRs) or short linear interacting motives (SLiMs), influence the formation and composition of the condensate [72, 73, 78]. These linear sequences encompass various interactions such as electrostatic interactions, π-π and cation-π contacts, hydrophobic interactions, and the valency and arrangement of LCRs [79,80,81,82] (see Fig. 1). However, the relevance of these sequences and the constraints governing their interactions are not yet fully understood [83,84,85]. Studies about the relationship between protein phase behavior and sequence modifications, such as deletions, truncations, or site-specific mutations, have revealed sequence-dependent characteristics that influence the phase separation of proteins [32, 33, 74, 86,87,88]. These include SLiMs and LCRs found within IDRs and modular interaction domains (MIDs) [32, 39]. MIDs are well-structured protein domains known for their essential functions in homo/heterotypic interactions among proteins, nucleic acids, or other molecules, i.e.: 14-3-3 domain, SH2 domain, Methyl-CpG DNA binding domain, etc. Unfortunately, the experimental challenges in studying IDRs limit our understanding of them. Recent discoveries highlight the pivotal roles of SLiMs, and LCRs in the formation of phase-separated condensates. SLiMs and LCRs, acting as mediators, play a crucial role in the selective partitioning and distinct composition of these condensates. By orchestrating such interactions, SLiMs and LCRs significantly contribute to shaping the architecture and functional diversity of cellular compartments, providing insights into the intricate mechanisms governing their formation and regulation [89]. Pappu and colleagues proposed the linkers-and-spacers model, which reduces this complexity to a pair of components: “linkers” as adhesive elements driving interactions, and “spacers” connecting stickers and influencing biomolecule-solvent interactions [78]. In IDRs, aromatic amino acids (Tyr or Phe) act as linkers, facilitating intra- and intermolecular contacts, while glycine and polar amino acids act as spacers without strong interaction patterns. Therefore, genetic variants on MIDs or IDRs can alter several aspects of BCs, including their formation, size, localization, material properties, and composition, consequently affecting the functional characteristics of BCs. This has been demonstrated in various pathologies, giving rise to the term “condensatopathies”: abnormal condensation leading to a specific disease phenotype [90] (see Fig. 1).
While only a subset of biomolecular components appears to be essential for maintaining condensate integrity [91,92,93], the potential number of these molecules within a condensate is vast, encompassing tens to hundreds of different biomolecules [94]. Thereby, condensates provide a platform for spatiotemporal regulation of cellular processes by self-organizing specific biomolecules and orchestrating their interactions [29].
Studying variant effects on condensate-promoting features like IDRs and their impact on BCs’ collective behavior can complement prioritization protocols, aiding in reclassifying VUS and enhancing diagnostics. Indeed, this approach may also provide valuable insights into phenotypic heterogeneity, missing heritability, and incomplete penetrance observed in RDs patients. We propose to analyze the effect of genetic variation in protein regions that promote condensation, such as IDRs, and their propensity to undergo phase separation due to the set of non-synonymous variants present in patients with RDs, using a single and multi-gene causation approach. These innovative strategies hold immense potential for identifying pathogenic variants, enhancing diagnostic capabilities for individuals affected by RDs, and elucidating their underlying molecular mechanisms, opening new avenues for therapeutic exploration.
To improve variant prioritization, we propose to study a new set of variables derived from in-silico predictors of disorder and condensation. We will assess the cumulative effect of patient-specific variants and their correlation with alterations in the composition of BCs, discriminating between linkers and spacers in scaffolds and clients. This comprehensive evaluation will elucidate the significance of individual components in disease manifestation and phenotypic diversity, providing deeper insights into the molecular underpinnings of disease and the relationship between genetic variations and phenotypes.
The method
To gain a deeper understanding of the role of BCs in cellular organization and function, it is essential to compile an accurate annotation of the IDRs of the human proteome, a precise inventory of those proteins involved in BCs, and all the competitive interactions among them. In the field of IDRs, the accumulation of experimental evidence over two decades has robustly substantiated the notion that IDRs can be inferred from sequence features. This body of research has paved the way for the development of databases and [95] multiple IDR prediction methods, employing diverse principles and sophisticated computing techniques [96, 97]. These advancements have significantly enhanced our capacity to identify and characterize IDRs, thus deepening our understanding of their functional significance in protein structure and function. The same has happened in the field of BCs, where research in the past decade has advanced in the elucidation of the protein composition of different BCs and the characterization of their roles as scaffolds and clients [98,99,100,101], thus allowing cataloging them according to their propensity to undergo condensation in-vitro[102,103,104,105,106].
In the evaluation of the propensity of each protein to condensate, it is noteworthy that while scaffolds have been recognized as pivotal components [107], clients, which do not possess inherent phase separation capabilities, may influence the formation and regulation of BCs through their interactions with one or more drivers [72, 108]. However, it should be noted that many clients have not been thoroughly characterized or individually tested in vitro, and the existence of additional scaffold proteins cannot be ruled out [109]. This distinction between scaffolds and clients underscores the challenge of predicting whether a protein will localize into a BC and whether changes in the aminoacidic composition will affect the cellular self-organizing process. While all BCs proteins may share certain standard features, those distinguishing clients from scaffolds differ. Therefore, further research is needed to unravel the precise mechanisms underlying protein localization to MLOs and to gain a more comprehensive understanding of its various properties.

Schematic representation of the workflow. WT P. Seqs, Wild type protein sequences; NSV P. Seqs, Non-synonymous variant derived protein sequences; RDr NSVPS, Rare disease related non-synonymous variant derived protein sequences; B, Benign; P, Pathological; VUS, Variant of uncertain significance; RD DBs, Rare diseases databases; BCs DBs, Protein-protein interactions, and biomolecular condensates databases;
Despite these weaknesses, depicted advances have greatly enhanced our understanding of the various factors influencing molecular condensation. Based on this knowledge, new computational methods have been developed to accurately predict the propensity of proteins to remain disordered or undergo condensation [73, 84, 110,111,112,113,114,115,116,117].
Machine learning algorithms enable us to explore the categories of the American College of Medical Genetics and the Association for Molecular Pathology [118] and others for predicting variant pathogenicity. Algorithms are trained on pathogenic and population variant data using a wide range of features including evolutionary information (such as “conserved sites”), gene-level properties (e.g., “essentiality”), and specific amino acid substitutions in protein sequences [119,120,121,122,123]. While these methods aid in predicting causality and improving genetic diagnosis [124, 125], predictions generated are not always biologically interpretable, making it difficult to determine the reasons why a particular missense variant is predicted to have a high or low pathogenicity score.
In this perspective, we propose to apply machine learning algorithms to the information from multiple predictors, network analysis metrics, and database annotation to enhance the classification of VUS, leading to more informed clinical decisions (see Fig. 2).
For model training and validation, variants from Clinvar [126] are segregated into a training dataset (containing well-characterized pathological and benign variants) and a test dataset (comprising likely benign, likely pathogenic, and VUS variants). Data preprocessing ensures data quality and reliability. For feature selection, we adopt a multi-faceted approach, incorporating predictors of IDRs to identify disorder propensity, linear interacting peptides, arginine and tyrosine-enriched domains, and polyproline regions within the protein sequence. By incorporating IDR predictors, such as MobiDB-Lite [127], flDPnn [128], and Bio2Bite tools (Disomine [129], Dynamine [130], Efoldmine [131], and Agmata [131, 132]) we predict protein biophysical properties from their amino-acid sequences. This enables us to capture the propensity of specific regions within a protein to exhibit disorder, thereby highlighting the potential impact of a variant on the protein-disordered regions. Secondly, to evaluate the likelihood of a protein undergoing phase separation and forming BCs, we apply condensation propensity predictors. These predictors leverage sequence features associated with condensate formation, such as LCRs, prion-like domains, and specific amino acid compositions. By employing established algorithms like ParSe [110], LLPhyScore [115], MaGS [133], PScore [134], and PhasePre [114], we assess the condensation propensity of proteins and identify variants that may influence their ability to form or modulate BCs. Condensation propensity predictors are used to evaluate the global and local likelihood of a protein and its regions undergoing phase separation. Third, in addition to sequence-based features, we incorporate topological measures derived from a bipartite protein-protein interaction network labeled as scaffolds and clients. By analyzing network properties such as degree centrality, betweenness centrality, nestedness, fuzzy modular segregation, and assortativity, we gain insights into the relevance of proteins within cellular processes and a comprehensive understanding of the functional relevance of genetic variants in the protein-protein interactions network. This enables us to identify variants that may disrupt critical protein interactions and perturb cellular pathways, thus providing valuable insights into the clinical significance of the variants. Finally, database information such as Scaffold or client annotation or HPO and GO terms related to the proteins is added to improve interpretability.
By integrating these sets of features, we aim to capture a wide range of biological characteristics associated with genetic variants. The inclusion of IDR predictors allows us to identify regions of disorder within proteins, highlighting their functional relevance. Condensation propensity predictors provide insights into the potential for phase separation and condensate formation, elucidating the role of variants in cellular organization. Topological measures derived from protein-protein interaction networks further enhance our understanding of the functional impact of these variants in cellular processes.
The comprehensive set of features selected in our proposed method facilitates a multi-dimensional analysis of genetic variants, enabling us to redefine their clinical significance. By leveraging machine learning algorithms, including support vector machines, random forests, and neural networks, to develop robust classification models with these informative features, we aim to develop a robust model for variant classification and provide a more accurate assessment of variants classified as VUSes. We hope that our approach will improve clinical decision-making and increase our understanding of the functional implications of genetic variants in the context of genetic diseases. These models are trained using a carefully selected training dataset over the features previously described and learn to classify VUSes as either pathogenic or benign, thus improving variant classification.
To prioritize informative features and maximize interpretability in machine learning models, we propose utilizing various strategies that offer valuable insights into the variant classification process. These strategies include feature importance analysis, partial dependence plots, individual instance interpretation, rule-based models, and model-agnostic interpretability techniques [135, 136].
Feature importance analysis reduces model dimensionality and prioritizes informative features, enabling clinicians to focus on critical factors driving accurate classification decisions. Partial dependence plots and individual instance interpretation techniques, visualize the relationship between specific variables and model output, allowing a clear understanding of their impact on variant classification independently of other variables. Individual instance interpretation techniques may provide detailed explanations for classifying individual variants, highlighting key factors considered by the model in its prediction. Model-agnostic interpretability techniques, such as LIME and SHAP, offer post-hoc explanations for any black-box machine learning model. By perturbing input features and analyzing the model’s response, these techniques generate local explanations that help clinicians understand the factors influencing predictions for individual variants. Finally, to enhance interpretability, rule-based models such as decision trees or rule sets could be employed. These models map input features to predicted classes, providing transparent guidelines for clinical decision-making.
By incorporating these interpretability methods, clinicians will gain access to a comprehensive toolbox for understanding and interpreting predictions made by the proposed machine-learning models. These strategies provide transparent insights into the decision-making process, instill trust in the model’s predictions, and facilitate effective integration into clinical practice. Examining these explanations will enable clinicians to validate and interpret the model’s predictions on a case-by-case basis, enhancing the overall utility of the models in clinical decision-making.
It won’t be an easy road
While the low frequency of each RD may seem insignificant for this type of study, in the US alone, approximately 30 million people are affected by RDs, impacting around 1 in 10 Americans [14]. Moreover, there are currently recognized between 5,000 and 10,000 RDs, depending on the source [137], providing a vast phenotypic landscape to explore the interdependence between variants and the unfolded phenotype.
Obviously, not all VUSes are linked to alterations in condensation processes. Exome sequencing covers less than 2% of the genome, allowing a diagnostic yield of around 30% [138] and leaving precise disease mechanisms largely unexplored [139]. Recent research has aimed at expanding the search space beyond coding regions to the immediate regulatory regions, revealing new pathogenic variants in a small fraction of cases [140]. Additionally, emerging reports suggest the involvement of distal enhancers and alterations in the three-dimensional (3D) genome structure in disease pathogenesis [141, 142]. Thus, the comprehensive exploration of the non-coding genome will provide valuable insights into the underlying mechanisms of genetic disorders and expand our understanding of the intricate regulatory networks that govern gene expression and cellular functions. In the context of the BCs these non-coding regions, whether expressed or not, have the potential to influence the cellular biomolecule composition. They can impact enhancers or promoter regions, alter the target selection of microRNAs, affect splicing variants, or influence transcript lifespan. Such changes can disrupt the critical balance of biomolecules involved in LLPS, thereby impacting condensation and the resulting biomolecular condensates’ composition. The reasons mentioned above further emphasize the necessity of adopting a systems biology approach. By integrating whole-genome sequencing, transcriptomic analysis, and computational models including biomolecular condensation propensity, competing RNA-RNA and RNA-protein interaction networks, and phenotypic enrichment, we can gain a comprehensive understanding of the underlying mechanisms of these diseases. This integrative approach could allow us to unravel the intricate interactions within biological systems and provide valuable insights into disease pathogenesis.
The fields of disorder and condensation prediction, as well as coarse-grained models of biomolecular self-organization, are rapidly evolving. However, it is important to note that predictors of disorder and condensation propensity, which rely on the primary sequence of proteins, have notable limitations [96, 117, 143]. The prediction of condensation propensity faces several challenges. For example, in the case of condensation propensity predictors, they commonly rely on a limited set of validated scaffolds for training algorithms, which greatly restricts their ability to accurately predict the condensation propensity of client proteins or other molecules also involved in the condensate. Additionally, our understanding of the underlying grammar of these processes is still very limited, and further experimental investigations are required to elucidate the logic behind condensation processes. Moreover, the role of post-translational modifications in triggering the condensation-decondensation process is well-known, but comprehensive data on these modifications for training machine learning algorithms are currently lacking. The prioritization of variants affecting linkers or spacers is a scientifically sound approach, provided the validity of the proposed model of linkers and spacers is acknowledged. It is crucial to recognize that models, albeit valuable tools, inherently reduce the complexity to achieve mathematical and computational tractability, potentially excluding critical information. Nevertheless, even with its limitations, employing a partial rule-based framework remains preferable to the absence of any guiding principles in variant prioritization.
Regarding disorder, the plasticity and interactivity of IDRs and their potential cellular function remain hard to predict [96, 127]. Understanding the grammar of IDRs is the first step on the path to deciphering these cellular self-organization processes. We lack experimental data about IDRs. However, given the experimental challenges associated with their study, multiple efforts are being made in the development of computational tools that enable us to delve deeper into this field, including the establishment of initiatives such as the Critical Assessment of Protein Intrinsic Disorder Prediction (CAPIDP) to set quality standards in the field. This highlights the continued interest in optimizing these predictors and in the need observed by the scientific community to access this valuable information for medical use.
Concluding remarks
In recent years, the concepts of intrinsically disordered regions IDRs, BCs, and liquid-liquid phase separation LLPS determinants have significantly advanced the fields of molecular biology and genetics, providing novel insights into gene regulation, protein function, and the underlying biology of diseases. As these concepts have matured, the integration of this knowledge into the prioritization of genetic variants becomes increasingly compelling. By incorporating predictors of functional properties of IDRs and condensation propensity in variant prioritization, we can take advantage of the mounting evidence that highlights the crucial role of condensation processes in disease pathogenesis. This integration promises to improve diagnostic accuracy, unravel molecular mechanisms underlying rare diseases, and facilitate the discovery of novel therapeutic targets and pathways, enabling innovative interventions for complex disorders. Combining advanced computational models with precision medicine approaches opens new horizons for more effective treatments, driving forward rare disease research and enhancing patient outcomes.
As the scientific understanding of IDRs, BCs, and LLPS continues to advance, their integration into clinical practice becomes increasingly essential. A comprehensive grasp of the complexities of genetic variant pathogenicity, including the impact of condensation processes, is crucial for improving diagnostic accuracy and patient care. Embracing this evolving field and incorporating predictive tools into clinical workflows better equips us to address the challenges posed by extensive genetic diversity, variable expressivity, and incomplete penetrance associated with rare diseases. Ultimately, integrating these cutting-edge approaches into clinical settings will lead to a more personalized and precise approach to medicine, yielding improved patient outcomes and deeper insights into genetic diseases.
Data availability
Abbreviations
- BCs:
-
Biomolecular condensates
- IDPs:
-
Intrinsically disordered proteins
- IDRs:
-
Intrinsically disordered regions
- LCRs:
-
Low complexity regions
- LLPS:
-
Liquid-liquid phase separation
- MIDs:
-
Modular interaction domains
- MLOs:
-
Membraneless organelles
- PTM:
-
Post-translational modification
- RDs:
-
Rare diseases
- SLiMs:
-
Short linear interacting motives
- VUS:
-
Variants of uncertain significance
References
Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. 2018;19(5):253–68.
Porubsky D, Vollger MR, Harvey WT, Rozanski AN, Ebert P, Hickey G et al. Gaps and complex structurally variant loci in phased genome assemblies. Genome Res [Internet]. 2023; https://doi.org/10.1101/gr.277334.122
Liao WW, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, et al. A draft human pangenome reference. Nature. 2023;617(7960):312–24.
Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, et al. Variant interpretation: functional assays to the rescue. Am J Hum Genet. 2017;101(3):315–25.
Hartman P, Beckman K, Silverstein K, Yohe S, Schomaker M, Henzler C, et al. Next generation sequencing for clinical diagnostics: five year experience of an academic laboratory. Mol Genet Metab Rep. 2019;19:100464.
Rouse SL, Florentine MM, Taketa E, Chan DK. Racial and ethnic disparities in genetic testing for hearing loss: a systematic review and synthesis. Hum Genet. 2022;141(3–4):485–94.
Samadder NJ, Riegert-Johnson D, Boardman L, Rhodes D, Wick M, Okuno S, et al. Comparison of Universal Genetic Testing vs Guideline-Directed targeted testing for patients with Hereditary Cancer Syndrome. JAMA Oncol. 2021;7(2):230–7.
Global Genes [Internet]. 2018 [cited 2023 Apr 5]. RARE Disease Facts. https://globalgenes.org/learn/rare-disease-facts/
McGuire AL, Gabriel S, Tishkoff SA, Wonkam A, Chakravarti A, Furlong EEM, et al. The road ahead in genetics and genomics. Nat Rev Genet. 2020;21(10):581–96.
Riordan JD, Nadeau JH. From peas to Disease: modifier genes, Network Resilience, and the Genetics of Health. Am J Hum Genet. 2017;101(2):177–91.
Ezquieta B, Santomé JL, Carcavilla A, Guillén-Navarro E, Pérez-Aytés A, Sánchez del Pozo J, et al. Alterations in RAS-MAPK genes in 200 Spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes. Genotype and cardiopathy. Rev Esp Cardiol. 2012;65(5):447–55.
Chen R, Shi L, Hakenberg J, Naughton B, Sklar P, Zhang J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe mendelian childhood diseases. Nat Biotechnol. 2016;34(5):531–8.
Bamshad MJ, Nickerson DA, Chong JX. Mendelian Gene Discovery: fast and furious with no end in Sight. Am J Hum Genet. 2019;105(3):448–55.
Global Genes [Internet]. 2022 [cited 2023 May 9]. Homepage. https://globalgenes.org/
Global Commission on Rare Disease [Internet]. [cited 2023 May 9]. https://www.globalrarediseasecommission.com/Report
Jacqueline I, Global G. 2014 [cited 2023 May 9]. Accurate Diagnosis of Rare Diseases Remains Difficult Despite Strong Physician Interest. https://globalgenes.org/blog/accurate-diagnosis-of-rare-diseases-remains-difficult-despite-strong-physician-interest-2/
Bauskis A, Strange C, Molster C, Fisher C. The diagnostic odyssey: insights from parents of children living with an undiagnosed condition. Orphanet J Rare Dis. 2022;17(1):233.
Fisher RA. XV.—the correlation between relatives on the supposition of mendelian inheritance. Trans R Soc Edinb. 1919;52(2):399–433.
Altenburg E, Muller HJ. The genetic basis of Truncate Wing,-an inconstant and modifiable character in Drosophila. Genetics. 1920;5(1):1–59.
Hivert V, Sidorenko J, Rohart F, Goddard ME, Yang J, Wray NR, et al. Estimation of non-additive genetic variance in human complex traits from a large sample of unrelated individuals. Am J Hum Genet. 2021;108(5):786–98.
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS Discovery: Biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–84.
Abdellaoui A, Yengo L, Verweij KJH, Visscher PM. 15 years of GWAS discovery: realizing the promise. Am J Hum Genet. 2023;110(2):179–94.
Boyle EA, Li YI, Pritchard JK. An expanded view of Complex traits: from polygenic to Omnigenic. Cell. 2017;169(7):1177–86.
Gjuvsland AB, Hayes BJ, Omholt SW, Carlborg Ö. Statistical epistasis is a generic feature of Gene Regulatory Networks. Genetics. 2007;175(1):411.
Domingo J, Baeza-Centurion P, Lehner B. The causes and consequences of genetic interactions (epistasis). Annu Rev Genomics Hum Genet. 2019;20:433–60.
Tsuchiya M, Giuliani A, Yoshikawa K. Cell-Fate Determination from Embryo to Cancer Development: Genomic Mechanism Elucidated. Int J Mol Sci [Internet]. 2020;21(13). https://doi.org/10.3390/ijms21134581
Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208.
Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–98.
Conti BA, Oppikofer M. Biomolecular condensates: new opportunities for drug discovery and RNA therapeutics. Trends Pharmacol Sci. 2022;43(10):820–37.
Sabari BR. Biomolecular condensates and Gene Activation in Development and Disease. Dev Cell. 2020;55(1):84–96.
Banani SF, Afeyan LK, Hawken SW, Henninger JE, Dall’Agnese A, Clark VE, et al. Genetic variation associated with condensate dysregulation in disease. Dev Cell. 2022;57(14):1776–e888.
Mensah MA, Niskanen H, Magalhaes AP, Basu S, Kircher M, Sczakiel HL, et al. Aberrant phase separation and nucleolar dysfunction in rare genetic diseases. Nature. 2023;614(7948):564–71.
Babu MM. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans. 2016;44(5):1185–200.
Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46.
Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–30.
Dunker AK, Babu MM, Barbar E, Blackledge M, Bondos SE, Dosztányi Z, et al. What’s in a name? Why these proteins are intrinsically disordered: why these proteins are intrinsically disordered. Intrinsically Disord Proteins. 2013;1(1):e24157.
Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–31.
Chong S, Mir M. Towards decoding the sequence-based Grammar governing the functions of intrinsically disordered protein regions. J Mol Biol. 2021;433(12):166724.
Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Protein disorder in the human diseasome: unfoldomics of human genetic diseases. BMC Genomics. 2009;10(Suppl 1):S12.
Tsang B, Pritišanac I, Scherer SW, Moses AM, Forman-Kay JD. Phase separation as a missing mechanism for interpretation of Disease mutations. Cell. 2020;183(7):1742–56.
Trivedi R, Nagarajaram HA. Intrinsically Disordered Proteins: An Overview. Int J Mol Sci [Internet]. 2022;23(22). https://doi.org/10.3390/ijms232214050
Perdigão N, Heinrich J, Stolte C, Sabir KS, Buckley MJ, Tabor B, et al. Unexpected features of the dark proteome. Proc Natl Acad Sci U S A. 2015;112(52):15898–903.
Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337(3):635–45.
Tompa P, Dosztanyi Z, Simon I. Prevalent structural disorder in E. Coli and S. Cerevisiae proteomes. J Proteome Res. 2006;5(8):1996–2000.
Mittal A, Holehouse AS, Cohan MC, Pappu RV. Sequence-to-conformation relationships of disordered regions tethered to folded domains of proteins. J Mol Biol. 2018;430(16):2403–21.
Choura M, Rebaï A. The disordered charged biased proteins in the human diseasome. Interdiscip Sci. 2020;12(1):44–9.
Vacic V, Markwick PRL, Oldfield CJ, Zhao X, Haynes C, Uversky VN, et al. Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput Biol. 2012;8(10):e1002709.
Coskuner-Weber O, Mirzanli O, Uversky VN. Intrinsically disordered proteins and proteins with intrinsically disordered regions in neurodegenerative diseases. Biophys Rev. 2022;14(3):679–707.
Khare SD, Chinchilla P, Baum J. Multifaceted interactions mediated by intrinsically disordered regions play key roles in alpha synuclein aggregation. Curr Opin Struct Biol. 2023;80:102579.
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by Disease Mutation. Cell. 2015;162(5):1066–77.
Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci U S A. 2006;103(22):8390–5.
Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, et al. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol Cell. 2012;46(6):871–83.
Buljan M, Chalancon G, Dunker AK, Bateman A, Balaji S, Fuxreiter M, et al. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr Opin Struct Biol. 2013;23(3):443–50.
Basu S, Bahadur RP. Conservation and coevolution determine evolvability of different classes of disordered residues in human intrinsically disordered proteins. Proteins. 2022;90(3):632–44.
Ahrens JB, Nunez-Castilla J, Siltberg-Liberles J. Evolution of intrinsic disorder in eukaryotic proteins. Cell Mol Life Sci. 2017;74(17):3163–74.
Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012;338(6114):1593–9.
Pajkos M, Mészáros B, Simon I, Dosztányi Z. Is there a biological cost of protein disorder? Analysis of cancer-associated mutations. Mol Biosyst. 2012;8(1):296–307.
Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science [Internet]. 2017;357(6357). https://doi.org/10.1126/science.aaf4382
Lyon AS, Peeples WB, Rosen MK. A framework for understanding the functions of biomolecular condensates across scales. Nat Rev Mol Cell Biol. 2021;22(3):215–35.
Hyman AA, Weber CA, Jülicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58.
Bergeron-Sandoval LP, Safaee N, Michnick SW. Mechanisms and consequences of Macromolecular phase separation. Cell. 2016;165(5):1067–79.
Klosin A, Oltsch F, Harmon T, Honigmann A, Jülicher F, Hyman AA, et al. Phase separation provides a mechanism to reduce noise in cells. Science. 2020;367(6476):464–8.
McSwiggen DT, Mir M, Darzacq X, Tjian R. Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 2019;33(23–24):1619–34.
Zhu G, Xie J, Kong W, Xie J, Li Y, Du L, et al. Phase separation of Disease-Associated SHP2 mutants underlies MAPK hyperactivation. Cell. 2020;183(2):490–e50218.
Taniue K, Akimitsu N. Aberrant phase separation and cancer. FEBS J. 2022;289(1):17–39.
Wang B, Zhang L, Dai T, Qin Z, Lu H, Zhang L, et al. Liquid-liquid phase separation in human health and diseases. Signal Transduct Target Ther. 2021;6(1):290.
Sawyer IA, Bartek J, Dundr M. Phase separated microenvironments inside the cell nucleus are linked to disease and regulate epigenetic state, transcription and RNA processing. Semin Cell Dev Biol. 2019;90:94–103.
Sanchez de Groot N, Torrent Burgas M, Ravarani CN, Trusina A, Ventura S, Babu MM. The fitness cost and benefit of phase-separated protein deposits. Mol Syst Biol. 2019;15(4):e8075.
Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, et al. Compositional control of phase-separated Cellular bodies. Cell. 2016;166(3):651–63.
Ditlev JA, Case LB, Rosen MK. Who’s in and who’s out-compositional control of Biomolecular condensates. J Mol Biol. 2018;430(23):4666–84.
Espinosa JR, Joseph JA, Sanchez-Burgos I, Garaizar A, Frenkel D, Collepardo-Guevara R. Liquid network connectivity regulates the stability and composition of biomolecular condensates with many components. Proc Natl Acad Sci U S A. 2020;117(24):13238–47.
Saar KL, Morgunov AS, Qi R, Arter WE, Krainer G, Lee AA et al. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc Natl Acad Sci U S A [Internet]. 2021;118(15). https://doi.org/10.1073/pnas.2019053118
Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, et al. A molecular Grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell. 2018;174(3):688–e9916.
Kilgore HR, Young RA. Learning the chemical grammar of biomolecular condensates. Nat Chem Biol. 2022;18(12):1298–306.
Chattaraj A, Blinov ML, Loew LM. The solubility product extends the buffering concept to heterotypic biomolecular condensates. Elife [Internet]. 2021;10. https://doi.org/10.7554/eLife.67176
Sabari BR, Dall’Agnese A, Young RA. Biomolecular condensates in the Nucleus. Trends Biochem Sci. 2020;45(11):961–77.
Choi JM, Holehouse AS, Pappu RV. Physical principles underlying the Complex Biology of Intracellular phase transitions. Annu Rev Biophys. 2020;49:107–33.
Gomes E, Shorter J. The molecular language of membraneless organelles. J Biol Chem. 2019;294(18):7115–27.
Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. 2015;11(11):899–904.
Dignon GL, Best RB, Mittal J. Biomolecular Phase separation: from Molecular Driving forces to Macroscopic Properties. Annu Rev Phys Chem. 2020;71:53–75.
Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. 2020;367(6478):694–9.
Vernon RM, Forman-Kay JD. First-generation predictors of biological protein phase separation. Curr Opin Struct Biol. 2019;58:88–96.
Chu X, Sun T, Li Q, Xu Y, Zhang Z, Lai L, et al. Prediction of liquid-liquid phase separating proteins using machine learning. BMC Bioinformatics. 2022;23(1):72.
Paiz EA, Allen JH, Correia JJ, Fitzkee NC, Hough LE, Whitten ST. Beta turn propensity and a model polymer scaling exponent identify intrinsically disordered phase-separating proteins. J Biol Chem. 2021;297(5):101343.
Li HR, Chiang WC, Chou PC, Wang WJ, Huang JR. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J Biol Chem. 2018;293(16):6090–8.
Elbaum-Garfinkle S, Kim Y, Szczepaniak K, Chen CCH, Eckmann CR, Myong S, et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci U S A. 2015;112(23):7189–94.
Dao TP, Kolaitis RM, Kim HJ, O’Donovan K, Martyniak B, Colicino E, et al. Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol Cell. 2018;69(6):965–e786.
Cermakova K, Hodges HC. Interaction modules that impart specificity to disordered protein. Trends Biochem Sci. 2023;48(5):477–90.
Mitrea DM, Mittasch M, Gomes BF, Klein IA, Murcko MA. Modulating biomolecular condensates: a novel approach to drug discovery. Nat Rev Drug Discov. 2022;21(11):841–62.
Monzon AM, Piovesan D, Fuxreiter M. Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates. Biomolecules [Internet]. 2022;12(1). https://doi.org/10.3390/biom12010092
Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, et al. Coexisting Liquid Phases Underlie Nucleolar Subcompartments Cell. 2016;165(7):1686–97.
Sanchez-Burgos I, Joseph JA, Collepardo-Guevara R, Espinosa JR. Size conservation emerges spontaneously in biomolecular condensates formed by scaffolds and surfactant clients. Sci Rep. 2021;11(1):15241.
Darling AL, Liu Y, Oldfield CJ, Uversky VN. Intrinsically disordered proteome of human membrane-less organelles. Proteomics. 2018;18(5–6):e1700193.
Piovesan D, Monzon AM, Quaglia F, Tosatto SCE. Databases for intrinsically disordered proteins. Acta Crystallogr D Struct Biol. 2022;78(Pt 2):144–51.
Necci M, Piovesan D, Predictors CAID, Curators DP, Tosatto SCE. Critical assessment of protein intrinsic disorder prediction. Nat Methods. 2021;18(5):472–81.
Basu S, Kihara D, Kurgan L. Computational prediction of disordered binding regions. Comput Struct Biotechnol J. 2023;21:1487–97.
Zhu M, Kuechler ER, Zhang J, Matalon O, Dubreuil B, Hofmann A et al. Proteomic analysis reveals the direct recruitment of intrinsically disordered regions to stress granules in. J Cell Sci [Internet]. 2020;133(13). https://doi.org/10.1242/jcs.244657
Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, et al. High-density proximity mapping reveals the Subcellular Organization of mRNA-Associated granules and bodies. Mol Cell. 2018;69(3):517–e3211.
Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, et al. Context-dependent and Disease-Specific Diversity in protein interactions within stress granules. Cell. 2018;172(3):590–e60413.
Woodruff JB, Ferreira Gomes B, Widlund PO, Mahamid J, Honigmann A, Hyman AA. The centrosome is a selective condensate that nucleates microtubules by concentrating Tubulin. Cell. 2017;169(6):1066–e7710.
Ning W, Guo Y, Lin S, Mei B, Wu Y, Jiang P, et al. DrLLPS: a data resource of liquid-liquid phase separation in eukaryotes. Nucleic Acids Res. 2020;48(D1):D288–95.
Mészáros B, Erdős G, Szabó B, Schád É, Tantos Á, Abukhairan R, et al. PhaSePro: the database of proteins driving liquid-liquid phase separation. Nucleic Acids Res. 2020;48(D1):D360–7.
You K, Huang Q, Yu C, Shen B, Sevilla C, Shi M, et al. PhaSepDB: a database of liquid-liquid phase separation related proteins. Nucleic Acids Res. 2020;48(D1):D354–9.
Hou C, Wang X, Xie H, Chen T, Zhu P, Xu X, et al. PhaSepDB in 2022: annotating phase separation-related proteins with droplet states, co-phase separation partners and other experimental information. Nucleic Acids Res. 2023;51(D1):D460–5.
Wang X, Zhou X, Yan Q, Liao S, Tang W, Xu P, et al. LLPSDB v2.0: an updated database of proteins undergoing liquid-liquid phase separation in vitro. Bioinformatics. 2022;38(7):2010–4.
Currie SL, Rosen MK. Using quantitative reconstitution to investigate multicomponent condensates. RNA. 2022;28(1):27–35.
Tejedor AR, Collepardo-Guevara R, Ramírez J, Espinosa JR. Time-Dependent Material properties of Aging Biomolecular condensates from different viscoelasticity measurements in Molecular Dynamics simulations. J Phys Chem B. 2023;127(20):4441–59.
Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid-liquid phase separation and Biomolecular condensates. Cell. 2019;176(3):419–34.
Ibrahim AY, Khaodeuanepheng NP, Amarasekara DL, Correia JJ, Lewis KA, Fitzkee NC, et al. Intrinsically disordered regions that drive phase separation form a robustly distinct protein class. J Biol Chem. 2023;299(1):102801.
Kuechler ER, Jacobson M, Mayor T, Gsponer J. GraPES: the granule protein Enrichment server for prediction of biological condensate constituents. Nucleic Acids Res. 2022;50(W1):W384–91.
Hardenberg M, Horvath A, Ambrus V, Fuxreiter M, Vendruscolo M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci U S A. 2020;117(52):33254–62.
van Mierlo G, Jansen JRG, Wang J, Poser I, van Heeringen SJ, Vermeulen M. Predicting protein condensate formation using machine learning. Cell Rep. 2021;34(5):108705.
Chen Z, Hou C, Wang L, Yu C, Chen T, Shen B, et al. Screening membraneless organelle participants with machine-learning models that integrate multimodal features. Proc Natl Acad Sci U S A. 2022;119(24):e2115369119.
Cai H, Vernon RM, Forman-Kay JD. An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions. Biomolecules [Internet]. 2022;12(8). https://doi.org/10.3390/biom12081131
Kuechler ER, Budzyńska PM, Bernardini JP, Gsponer J, Mayor T. Distinct features of stress granule proteins predict localization in Membraneless Organelles. J Mol Biol. 2020;432(7):2349–68.
Vendruscolo M, Fuxreiter M. Towards sequence-based principles for protein phase separation predictions. Curr Opin Chem Biol. 2023;75:102317.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Nicora G, Zucca S, Limongelli I, Bellazzi R, Magni P. A machine learning approach based on ACMG/AMP guidelines for genomic variant classification and prioritization. Sci Rep. 2022;12(1):2517.
McInnes G, Sharo AG, Koleske ML, Brown JEH, Norstad M, Adhikari AN, et al. Opportunities and challenges for the computational interpretation of rare variation in clinically important genes. Am J Hum Genet. 2021;108(4):535–48.
Quinodoz M, Peter VG, Cisarova K, Royer-Bertrand B, Stenson PD, Cooper DN, et al. Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am J Hum Genet. 2022;109(3):457–70.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Baugh EH, Simmons-Edler R, Müller CL, Alford RF, Volfovsky N, Lash AE, et al. Robust classification of protein variation using structural modelling and large-scale data integration. Nucleic Acids Res. 2016;44(6):2501–13.
Li Q, Zhao K, Bustamante CD, Ma X, Wong WH. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet Med. 2019;21(9):2126–34.
Banda JM, Sarraju A, Abbasi F, Parizo J, Pariani M, Ison H, et al. Finding missed cases of familial hypercholesterolemia in health systems using machine learning. NPJ Digit Med. 2019;2:23.
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48(D1):D835–44.
Necci M, Piovesan D, Clementel D, Dosztányi Z, Tosatto SCE. MobiDB-lite 3.0: fast consensus annotation of intrinsic disorder flavors in proteins. Bioinformatics. 2021;36(22–23):5533–4.
Hu G, Katuwawala A, Wang K, Wu Z, Ghadermarzi S, Gao J, et al. flDPnn: accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat Commun. 2021;12(1):4438.
Orlando G, Raimondi D, Codicè F, Tabaro F, Vranken W. Prediction of disordered regions in proteins with recurrent neural networks and Protein dynamics. J Mol Biol. 2022;434(12):167579.
Cilia E, Pancsa R, Tompa P, Lenaerts T, Vranken WF. From protein sequence to dynamics and disorder with DynaMine. Nat Commun. 2013;4:2741.
Raimondi D, Orlando G, Pancsa R, Khan T, Vranken WF. Exploring the sequence-based prediction of folding initiation sites in proteins. Sci Rep. 2017;7(1):8826.
Orlando G, Silva A, Macedo-Ribeiro S, Raimondi D, Vranken W. Accurate prediction of protein beta-aggregation with generalized statistical potentials. Bioinformatics. 2020;36(7):2076–81.
Farahi N, Lazar T, Wodak SJ, Tompa P, Pancsa R. Integration of Data from Liquid-Liquid Phase Separation Databases Highlights Concentration and Dosage Sensitivity of LLPS Drivers. Int J Mol Sci [Internet]. 2021;22(6). https://doi.org/10.3390/ijms22063017
Vernon RM, Chong PA, Tsang B, Kim TH, Bah A, Farber P et al. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife [Internet]. 2018;7. https://doi.org/10.7554/eLife.31486
Murdoch WJ, Singh C, Kumbier K, Abbasi-Asl R, Yu B. Definitions, methods, and applications in interpretable machine learning. Proc Natl Acad Sci U S A. 2019;116(44):22071–80.
Nandi A, Pal AK. Interpreting machine learning models: learn Model Interpretability and Explainability methods. A; 2021. p. 343.
Haendel M, Vasilevsky N, Unni D, Bologa C, Harris N, Rehm H, et al. How many rare diseases are there? Nat Rev Drug Discov. 2020;19(2):77–8.
Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97(2):199–215.
Rehm HL. Evolving health care through personal genomics. Nat Rev Genet. 2017;18(4):259–67.
Pena LDM, Jiang YH, Schoch K, Spillmann RC, Walley N, Stong N, et al. Looking beyond the exome: a phenotype-first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet Med. 2018;20(4):464–9.
Xu J, Song F, Lyu H, Kobayashi M, Zhang B, Zhao Z, et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature. 2022;611(7935):387–98.
Spielmann M, Lupiáñez DG, Mundlos S. Structural variation in the 3D genome. Nat Rev Genet. 2018;19(7):453–67.
Kuechler ER, Huang A, Bui JM, Mayor T, Gsponer J. Comparison of Biomolecular Condensate Localization and Protein Phase Separation Predictors. Biomolecules [Internet]. 2023;13(3). https://doi.org/10.3390/biom13030527
Acknowledgements
We are deeply grateful to all those who played a role in the success of this project. We would like to thank Begoña Puga Lopez for their invaluable input and human support throughout the research process.
Funding
Funding for open access publishing: Universidad de Cádiz/CBUA. This research was funded by the Andalusian Ministry of Health and Families through the initiative (RPS 24664), code PI-0069-2022; from group BIO267 and CTS927 (Andalusian Government), and by grants PID2022-138181OB-I00 (MICINN and FEDER). The “CIBER de Enfermedades Raras” is an initiative from the ISCIII (Spain).
Funding for open access publishing: Universidad de Cádiz/CBUA
Author information
Authors and Affiliations
Contributions
MHT, RM, and AMLS conceived of the presented idea. MHT and RM were major contributors in writing the manuscript. RM designed the figures. AMLS and MAM obtained funding and supervised the project. All authors discussed the results and worked and commented on the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Heredia-Torrejón, M., Montañez, R., González-Meneses, A. et al. VUS next in rare diseases? Deciphering genetic determinants of biomolecular condensation. Orphanet J Rare Dis 19, 327 (2024). https://doi.org/10.1186/s13023-024-03307-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-024-03307-6