
Abstract
Background
Premature ovarian insufficiency (POI) is a highly heterogeneous disease, and up to 25% of the cases can be explained by genetic causes. G protein-coupled receptor 3 (GPR3) plays an important role in oocyte arrest, and Gpr3-deficient mice exhibited POI-like phenotypes.
Case presentation
We identified two heterozygous missense variants of GPR3: NM_005281: c.C973T (p.R325C) and c.G772A (p.A258T) in two sporadic Han Chinese POI cases through whole exome sequencing and genetic analysis. The two patients were diagnosed as POI in their late 20s, presenting elevated serum levels of follicle stimulating hormone and secondary amenorrhea. Both variants are very rare in the population databases of ExAC, gnomAD and PGG.Han. The affected amino acids are conserved across species and the mutated amino acids are predicted deleterious with bioinformatics prediction tools and the protein three-dimensional structure analysis.
Conclusions
It is the first report of rare GPR3 variants associated with POI women, providing an important piece of evidence for GPR3 as a candidate gene which should be screened in POI. This finding suggested the necessity of including GPR3 in etiology study and genetic counseling of POI patients.
Similar content being viewed by others


Background
Premature ovarian insufficiency (POI) is a common female reproductive disease characterized by menstrual disturbance (amenorrhea or oligomenorrhea) combined with elevated serum follicle stimulating hormone (FSH) level (> 25 IU/L) before 40 years of age [1]. A recent study showed that the prevalence of POI is 3.7% in women [2]. Due to the reduced ovarian function, POI patients suffer from menstrual disorders and fertility difficulties, and they also display higher likelihoods of developing many other long-term physical conditions and mental diseases than normal women.
The causative factors of POI are highly heterogeneous, including genetic factors, autoimmune diseases, iatrogenic injuries and among others. It is estimated that genetic factors account for approximately 25% of POI patients [3]. Especially in recent years, many pathogenic genetic variants including single nucleotide variants (SNVs), small-scale insertions and deletions (Indels) and copy number variations (CNVs) have been characterized in familial or sporadic POI patients with the usage of high-throughput sequencing technology [4,5,6]. These variants are found to locate in genes mainly involved in meiosis, DNA damage response, steroidogenesis, follicle activation and development, etc. [5].
The human G protein-coupled receptor 3 (GPR3), located on 1p36.11, is predominantly expressed in oocytes, testis and brain [7]. GPR3 (NM_005281) sencodes a protein of 330 amino acids with seven transmembrane domains, which belongs to the G protein-coupled receptor family and can activate Gs protein and elevate cAMP levels in oocytes [7, 8]. GPR3 has been identified as a crucial factor in the maintenance of meiotic arrest and maturation in oocytes through ex vivo studies [9,10,11]. Female mice that lack Gpr3 develop premature ovarian aging due to spontaneous resumption of meiosis in antral follicles, exhibiting reduced fertility in young age and severe infertility in old age [7, 12]. These results altogether suggested GPR3 as an attractive candidate gene of human POI. Several studies therefore tried to identify disease-associated variants of GPR3 in POI patients, however all the results were disappointing [13,14,15]. Whether GPR3 could be classified as a candidate gene of POI has become a concern. In the present study, our cohort comprises 156 Han Chinese women with sporadic POI as previously described [16, 17]. Two rare missense variants in GPR3 were identified in two cases.
Case presentation
Study participants
One hundred fifty six Chinese women with sporadic POI have been included in our study at the Obstetrics and Gynecology Hospital of Fudan University between February 2017 and March 2022. The informed consent was obtained from all participants. The inclusion criteria consisted of amenorrhea for at least 4 months before 40 years of age and two serum FSH levels greater than 25 IU/L with intervals of more than 4 weeks. Women with ovarian surgery or radiotherapeutic or chemotherapeutic interventions were excluded. Genomic DNA was extracted from peripheral blood samples using the QIAamp DNA Blood Mini Kit (QIAGEN).
Whole exome sequencing (WES) and data processing
Genomic DNA was then subjected to WES at iGeneTech Bioscience (Beijing, China). Raw data were mapped to the human reference genome sequence (GRCh37/hg19) using the Burrows-Wheeler Alignment tool. The Genome Analysis Toolkit was used to accomplish the variant calling. All variants were further annotated with ANNOVAR software. The processing of genetic analysis was as previously described [17]. Briefly, the medium and high-quality variations were retained, and then the genetic variants in the exonic and splicing regions were chosen. Variant filtering was performed based on a minor allele frequency (MAF) ≤ 1‰ in population databases of the Exome Aggregation Consortium (ExAC), the Genome Aggregation Database (gnomAD) and the Han Chinese Genomes Database (PGG.Han). Synonymous mutations were filtered out. After searching for GPR3 variants, only two missense variants located in GPR3 (NM_005281) were identified and then they were investigated by SIFT, MutationTaster, CADD and DANN for further analysis. Finally, all of these two GPR3 variants carried by F057 and F086 respectively predicted to be deleterious were retained.
Clinical case report
Clinical information of F057 and F086 are summarized in Table 1. There was no history of any ovarian surgery, uterine surgery, radiotherapy, chemotherapy or autoimmune thyroid disease in the two patients.
F057 had normal physical development and had spontaneous menarche at the age of 14 with regular cycle. Her menstrual cycle became irregular when she was 26 years old. She was diagnosed with POI one year later and was administered with femoston for maintenance of menstruation. As shown in Table 1, her hormone examination exhibited an increased level of FSH (68.34 IU/L) and a rather low AMH level (0.01 ng/mL). Ultrasound examination results showed she had relatively small ovarian volume, without any obvious follicles on both sides.
F086 reported regular pubertal development with spontaneous menarche at 14 years of age, but the length of her menstrual cycle is about 20 days and she had severe menorrhagia. Her irregular menstruation became even worse after the age of 19 and she was diagnosed with POI at the age of 29. The results of hormone evaluation showed an elevated level of FSH (114.63 IU/L) and a decreased E2 concentration (17.08 pg/mL). Ultrasound examination displayed that she also had small ovaries without any obvious follicles on both sides.
Identification of GPR3 variants
Two different rare variations in the coding region of GPR3 were identified in F057 and F086 respectively (Table 2). The authenticity of two variants was first confirmed by Sanger sequencing (Fig. 1A). However, F057’s parental samples were unavailable. Sanger sequencing revealed that F086's variation was inherited from her mother, who had irregular menstruation from the age of 21, followed by sporadic menstruation.

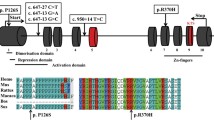
Identification of GPR3 variants in two POI patients. A Sanger sequencing confirmed the heterozygous GPR3 variants in patients. The red arrows indicate the positions of the two variants. B The amino acid residues of GPR3 corresponding to the identified variants are conserved across species. The red arrows indicate the positions of the affected residues. C Overview of the predicted structure of GPR3 protein. The enlarged boxes indicate the locations of the affected residues
F057 was found to carry a heterozygous GPR3 c.G772A (p.A258T) variant and F086 carried a heterozygous GPR3 c.C973T (p.R325C) variant. Both variants have low frequencies in the ExAC and gnomAD population databases and are absent in the PGG.Han database, which includes genomic data of 114,783 Han Chinese individuals [18]. The c.G772A (p.A258T) variant was predicted to be deleterious by SIFT, MutationTaster and DANN, while the c.C973T (p.R325C) variant was harmful according to all mutation assessments (Table 2).
Additionally, both variants are highly evolutionarily conserved across species (Fig. 1B). According to the three-dimensional structure of wild-type GPR3 protein obtained from the Uniprot database (Uniprot entry: P46089), it is found that Ala258 was located in the sixth transmembrane domain (Fig. 1C) and the p.A258T variant may affect the protein structure. Additionally, the Arg325 was located in intracellular surface and the change from arginine to cysteine could affect the interaction with Ser324, which may lead to dysregulation of intracellular cAMP signaling. In addition, according to 3D structure prediction, Arg325 also interact with Glu302 at the end of the seventh transmembrane domain, thus p.R325C variant may also affect this interaction and the whole protein structure.
Literature review
We searched the PubMed database using text words relating to “GPR3” and “POI” or “oocyte” and summarized the representative supporting evidence for GPR3 as a candidate gene for POI (Table 3). Previous clinical studies of GRP3 variants in POI patients have also been reviewed.
GPR3 was first characterized as a pivotal factor for oocyte meiosis in 2004, when Mehlmann et al. found that GPR3 maintained the prophase I arrest in mice [7]. Most oocytes from Gpr3 knockout mouse resumed meiosis within antral follicles spontaneously. Ledent et al. found that Gpr3 deficient mice were subfertile and further demonstrated that Gpr3 protected and possibly rescued oocytes from aging [12]. He also found increased oocyte fragmentation in superovulated Gpr3−/− female mice, suggesting a higher proportion of atresia and poor meiotic oocytes. In addition, Gpr3 knockout mice were found to have POI-like phenotypes including increased FSH levels and shorter estrus cycles [12]. Phenotypes in Gpr3 knockout mice were also mimicked by specific reduction of Gpr3 in the mouse oocyte using RNAi techniques [9]. Oocytes injected with Gpr3 siRNA lost their ability to maintain meiotic arrest and the GVBD rate observed was very close to the rate seen in Gpr3 global knockout mice. Similar experimental results were obtained in porcine oocytes [11]. Injection of siRNA targeting GPR3 stimulated meiotic resumption of oocytes, while overexpression of GPR3 inhibited meiotic maturation of porcine oocytes. Mechanism study further found that Gpr3 was expressed and remained active before cAMP is required to maintain meiotic arrest, indicating another role of Gpr3 in the acquisition of oocyte meiotic competence [19]. Taken together, these functional studies and animal models all suggested GPR3 as an attractive candidate gene for human POI.
However, when Ertug Kovanci et al.first tried to find any sequence variants in 82 American premature ovarian failure (POF) women, he failed to detect any potentially disease-associated GPR3 variants among them [13]. Similarly, no GPR3 mutations affecting the coding sequence was found in 100 Chinese POF patients in 2010 [14]. In a recent study, 269 POI patients were screened for several causative/candidate POI genes including GPR3 by Ion Torrent semiconductor sequencing and none of them presented any pathogenic variants of GPR3 [15]. Therefore, our study is the first report to identify rare and likely pathogenic variants of GPR3 in two Chinese POI patients, presenting evidence that GPR3 is involved in POI pathogenesis.
Discussion and conclusions
Identifying causative variants in POI patients has been challenging with the high heterogeneity in etiology. Although 159 POI-related genes have been listed in the Ovarian Kaleidoscope database (OKdb), the genetic causes remain to be elucidated in the majority of clinical POI patients [5]. Herein, we identified two rare GPR3 variants in a cohort of 156 sporadic POI patients, and the harmfulness of them were further confirmed by bioinformatics software. To exclude the potential contribution of other pathogenic variants in F057 and F086, we performed the genetic analysis on all the variants from the two patients. As shown in the supplementary Table 1, after a series of filtering steps, there was no rare and deleterious variant of any other POI causative/candidate genes except GPR3 in both patients. However, considering that the genetic etiological spectrum of POI is still expanding, it is undeniable that there is the possibility that other novel POI causative/candidate genes might involve in the pathogenesis of POI.
POI is a complex disorder with high genetic heterogeneity. No POI-related gene is implicated in more than 5% of sporadic cases, and most POI genes could not be replicated in different reports. For example, FMR1 premutation is one of the most common causes of POI in western countries. Around 11–14% of familial and 2–6% of sporadic POI cases are associated with FMR1 premutation [20]. However, there is a rather lower contribution (< 1%) of FMR1 premutation in Chinese POI women [21,22,23]. More recently, a large cohort of 375 POI patients have been subjected to genetic analysis by targeted sequencing (88 POI-related genes) and whole exome sequencing [24]. Pathogenic variants in 50 POI genes were detected, most of which were identified in only one patient. More importantly, no variant was identified in the other POI genes at all. Taken all these together, we think that ethnicity difference, limited sample size and genetic heterogeneity of POI may also affect the contribution of GPR3 variants in different POI studies.
GPR3 is predominantly expressed in oocytes [7]. Studies with genetic or oligonucleotide-mediated ablation of GPR3 in mice provided supporting evidence that GPR3 is required to maintain high cAMP levels in the oocytes and meiotic arrest [7, 9]. GPR3 is also expressed in Xenopus, porcine and human oocytes, suggesting a conserved function across species [10, 11, 25]. A phenotype reminiscent of human premature ovarian failure was also reported in GPR3 deficient mice by Ledent et al. [12]. Resumption of meiosis in Gpr3 siRNA-injected oocytes was dose-dependent [9], suggesting that the amount of Gpr3 is important. It also suggests a possibility for the pathogenicity of heterozygous deleterious variants of GPR3. However, the molecular mechanism of GPR3 affecting meiosis is still unclear, whether there are undiscovered protein mediators in the related pathway needs further study. Moreover, the pathogenesis of GPR3 variation identified in this work remained to be further elucidated in future by molecular methods or animal models.
In conclusion, we reported for the first time two rare variants of GPR3 associated with POI patients, providing important evidence for GPR3 as a candidate gene that should be screened for in POI patients. Several previous reports have not found any GPR3 variation in POI patients, probably due to the high heterogeneity of POI in etiology. A larger cohort of POI patients is expected to precisely assess the contribution of GPR3 variation in POI and to define the genotype–phenotype correlations in women carrying GPR3 variants.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AMH:
-
Anti-Müllerian hormone
- CNV:
-
Copy number variation
- DHPLC:
-
Denaturing high-performance liquid chromatography
- E2:
-
Estradiol
- ExAC:
-
Exome Aggregation Consortium
- FSH:
-
Follicle stimulating hormone
- gnomAD:
-
Genome Aggregation Database
- GPR3:
-
G protein-coupled receptor 3
- LH:
-
Luteinizing hormone
- MAF:
-
Minor allele frequency
- OKdb:
-
Ovarian Kaleidoscope database
- P:
-
Progesterone
- PCR:
-
Polymerase chain reaction
- PGG.Han:
-
Han Chinese Genomes Database
- POF:
-
Premature ovarian failure
- POI:
-
Premature ovarian insufficiency
- PRL:
-
Prolactin
- SNV:
-
Single nucleotide variant
- T:
-
Testosterone
- WES:
-
Whole-exome sequencing
References
European Society for Human Reproduction and Embryology (ESHRE) Guideline Group on POI, Webber L, Davies M, Anderson R, Bartlett J, et al. ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016;31(5):926–37.
Golezar S, Ramezani TF, Khazaei S, Ebadi A, Keshavarz Z. The global prevalence of primary ovarian insufficiency and early menopause: a meta-analysis. Climacteric. 2019;22(4):403–11.
Qin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update. 2015;21(6):787–808.
Jiao X, Ke H, Qin Y, Chen ZJ. Molecular genetics of premature ovarian insufficiency. Trends Endocrinol Metab. 2018;29(11):795–807.
Yang Q, Mumusoglu S, Qin Y, Sun Y, Hsueh AJ. A kaleidoscopic view of ovarian genes associated with premature ovarian insufficiency and senescence. FASEB J. 2021;35(8): e21753.
Franca MM, Mendonca BB. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract Res Clin Endocrinol Metab. 2022;36(1): 101594.
Mehlmann LM, Saeki Y, Tanaka S, Brennan TJ, Evsikov AV, et al. The Gs-linked receptor GPR3 maintains meiotic arrest in mammalian oocytes. Science. 2004;306(5703):1947–50.
Freudzon L, Norris RP, Hand AR, Tanaka S, Saeki Y, et al. Regulation of meiotic prophase arrest in mouse oocytes by GPR3, a constitutive activator of the Gs G protein. J Cell Biol. 2005;171(2):255–65.
Mehlmann LM. Oocyte-specific expression of Gpr3 is required for the maintenance of meiotic arrest in mouse oocytes. Dev Biol. 2005;288(2):397–404.
Deng J, Lang S, Wylie C, Hammes SR. The Xenopus laevis isoform of G protein-coupled receptor 3 (GPR3) is a constitutively active cell surface receptor that participates in maintaining meiotic arrest in X. Laevis Oocytes Mol Endocrinol. 2008;22(8):1853–65.
Yang CR, Wei YC, Qi ST, Chen L, Zhang QH, et al. The G protein coupled receptor 3 is involved in cAMP and cGMP signaling and maintenance of meiotic arrest in porcine oocytes. PLoS ONE. 2012;7(6): e38807.
Ledent C, Demeestere I, Blum D, Petermans J, Hamalainen T, et al. Premature ovarian aging in mice deficient for Gpr3. Proc Natl Acad Sci U S A. 2005;102(25):8922–6.
Kovanci E, Simpson JL, Amato P, Rohozinski J, Heard MJ, et al. Oocyte-specific G-protein-coupled receptor 3 (GPR3): no perturbations found in 82 women with premature ovarian failure (first report). Fertil Steril. 2008;90(4):1269–71.
Zhou S, Wang B, Ni F, Wang J, Cao Y, et al. GPR3 may not be a potential candidate gene for premature ovarian failure. Reprod Biomed Online. 2010;20(1):53–5.
Eskenazi S, Bachelot A, Hugon-Rodin J, Plu-Bureau G, Gompel A, et al. Next generation sequencing should be proposed to every woman with “Idiopathic” primary ovarian insufficiency. J Endocr Soc. 2021;5(7):bvab032.
Yang X, Zhang X, Jiao J, Zhang F, Pan Y, et al. Rare variants in FANCA induce premature ovarian insufficiency. Hum Genet. 2019;138(11–12):1227–36.
Li G, Yang X, Wang L, Pan Y, Chen S, et al. Haploinsufficiency in non-homologous end joining factor 1 induces ovarian dysfunction in humans and mice. J Med Genet. 2022;59(6):579–88.
Gao Y, Zhang C, Yuan L, Ling Y, Wang X, et al. PGG.Han: the Han Chinese genome database and analysis platform. Nucleic Acids Res. 2020;48(D1):D971–6.
Firmani LD, Uliasz TF, Mehlmann LM. The switch from cAMP-independent to cAMP-dependent arrest of meiotic prophase is associated with coordinated GPR3 and CDK1 expression in mouse oocytes. Dev Biol. 2018;434(1):196–205.
Man L, Lekovich J, Rosenwaks Z, Gerhardt J. Fragile X-associated diminished ovarian reserve and primary ovarian insufficiency from molecular mechanisms to clinical manifestations. Front Mol Neurosci. 2017;10:290.
Guo T, Qin Y, Jiao X, Li G, Simpson JL, et al. FMR1 premutation is an uncommon explanation for premature ovarian failure in Han Chinese. PLoS One. 2014;9(7):e103316.
Ye Y, Lan X, Cong J, Li N, Wu Y, et al. Analysis of CGG repeats in FMR1 in Chinese women with idiopathic premature ovarian failure. Reprod Biomed Online. 2014;29(3):382–7.
Lu C, Li R, Chen X, Xu Y, Yan L, et al. The “normal” range of FMR1 triple CGG repeats may be associated with primary ovarian insufficiency in China. Reprod Biomed Online. 2017;34(2):175–80.
Heddar A, Ogur C, Da Costa S, Braham I, Billaud-Rist L, et al. Genetic landscape of a large cohort of Primary Ovarian Insufficiency: New genes and pathways and implications for personalized medicine. EBioMedicine. 2022;84: 104246.
DiLuigi A, Weitzman VN, Pace MC, Siano LJ, Maier D, et al. Meiotic arrest in human oocytes is maintained by a Gs signaling pathway. Biol Reprod. 2008;78(4):667–72.
Acknowledgements
The authors gratefully thank the patients for participating and supporting this study.
Funding
This work was supported by National Key Research and Development Program of China (2022YFC2703800) and National Natural Science Foundation of China (32270658 and 32288101).
Author information
Authors and Affiliations
Contributions
SR, XZ and YW designed the study. LS and XZ provided patients’data and performed clinical assessments. SR, FZ, LS, XY, YP, and YW analyzed data. SR conducted experiments and wrote the manuscript. YW edited the manuscript. FZ, XZ and YW supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Declaration of Helsinki and approved by the institutional review boards of the centers participating in this study.
Consent for publication
All patients gave their signed informed consent for this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary table 1.
Filtering steps of two WES data processing. Supplementary table 2. Primers used for Sanger sequencing. Supplementary table 3. List of 101 known POI causative genes. Supplementary table 4. List of 92 POI candidate genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ren, S., Zhang, F., Shang, L. et al. Rare variants in GPR3 in POI patients: a case series with review of literature. J Ovarian Res 16, 210 (2023). https://doi.org/10.1186/s13048-023-01282-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-023-01282-3