
Abstract
Epigenetic (“above genetics”) modifications can alter the gene expression without altering the DNA sequence. Aberrant epigenetic regulations in cancer include DNA methylation, histone methylation, histone acetylation, non-coding RNA, and mRNA methylation. Epigenetic-targeted agents have demonstrated clinical activities in hematological malignancies and therapeutic potential in solid tumors. In this review, we describe mechanisms of various epigenetic modifications, discuss the Food and Drug Administration-approved epigenetic agents, and focus on the current clinical investigations of novel epigenetic monotherapies and combination therapies in solid tumors.
Similar content being viewed by others

Background
Carcinogenesis is a complex process that involves both genetic and epigenetic changes, leading to the transformation of normal cells into malignant cells. The aberrant genetic and epigenetic alterations are the hallmark of cancer. Epigenetic modifications are responsible for cellular plasticity, differentiation and reprogramming without altering the underlying DNA sequence of the organism [1]. Normal cell development depends on regulated transcription of critical proteins, and individual cells within specific tissues and organs maintain their unique biological functions based on heritable and evolutionary differences in the DNA packaging. Histone proteins (two copies of histones H2A, H2B, H3 and H4) wrap around 147 base pairs of DNA to form a nucleosome. Nucleosomes are further compacted by additional proteins to form chromatin. Epigenetic modifications, including acetylation and methylation (histone marks), can alter DNA accessibility and chromatin structure and regulate gene transcription activation or silencing. Acetylated histones are less compact, thereby enabling gene transcription by making the DNA more accessible to RNA polymerase and the transcriptional machinery. On the other end, methylated histones can be either repressive or activating, depending on the site and degree of methylation. Methylation of histone H3 at lysine 4, 36 and 79 is generally considered as an activation mark, whereas methylations on histone H3 lysine 9, 27 are linked to transcriptional repression [2]. In general, enzymes that add acetyl or methyl groups to the histone or DNA are referred to as “writers”, whereas enzymes that remove histone marks are called “erasers”. Proteins that recognize histone and DNA modifications are the chromatin “readers” [1].
The complex balance of normal and abnormal epigenetic regulation is an area of intense interest in cancer research, including therapeutic development in cancer [3]. This article will illustrate aberrant changes in DNA methylation, histone acetylation and histone methylation (summarized in Fig. 1) in cancer, discuss the epigenetic agents in both hematological malignancies and solid tumors, and highlight the recent novel combination strategies, such as with immune checkpoint inhibitors and hormonal therapies, in solid tumors.

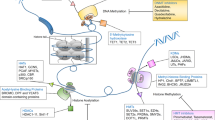
The epigenetic readers, writers and erasers. (a) Histone proteins wrap around DNA to form a nucleosome, which are then compacted to form chromatins and further into chromosomes. HATs add acetyl groups and HDACs remove acetyl groups from histone lysine residues. The acetylated histones are considered as “open chromatin”, enabling gene transcription, whereas deacetylated histones are “closed chromatin” and associated with gene silencing. BET proteins recognize acetylated histones and are involved with transcriptional activation by recruiting other proteins. In comparison with histone acetylation, histone methylation can be either repressive or activating, depending on the site and degree of methylation. Different histone methyltransferases are specific to modify the lysine or arginine residues. LSD1 demethylates either the active mark of H3K4 or the repressive mark of H3K9, in a context-dependent manner. EZH2 methylates H3K27 and promotes transcription silencing. DOT1L methylates H3K79, which is an activation mark. At the DNA level, DNMTs methylate and convert cytosine to 5-methylcytosine (5mC), and TETs remove methyl groups on DNA. Mutations in genes encoding enzymes in the cellular metabolism can alter the epigenetic landscape. This is exemplified by IDH1/2 that metabolize isocitrate to α-KG. IDH1/2 mutations (gain-of-function) result in further processing of α-KG to 2-HG (“oncometabolite”), which inhibits TETs and leads to reduced DNA demethylation (increased DNA methylation state). b A multiprotein complex (consisting METTL3, METTL14 and other subunits) methylates adenosine base at the nitrogen-6 position and forms m6A in the messenger RNA. m6A modification is reversible and it can be erased by ALKBH5 and FTO. m6A reader proteins can regulate the metabolism of mRNA. For example, YTHDF2 binds to m6A and targets mRNA degradation. HAT histone acetyltransferase, HDAC histone deacetylase, BET bromodomain and extra-terminal motif proteins, LSD1 lysine-specific histone demethylase 1, EZH2 enhancer of zeste homolog 2, DOT1L disruptor of telomeric silencing 1 like, DNMT DNA methyltransferase, TET ten-eleven translocation, IDH isocitrate dehydrogenase, α-KG α-ketoglutarate, 2-HG 2-hydroxyglutarate, m6A N6-methyladenosine, METTL3 methyltransferase-like protein 3, METTL14 methyltransferase-like protein 14, ALKBH5 alkB homolog 5, FTO fat-mass and obesity associated protein
Main text
Therapeutics targeting the cancer epigenome
Therapeutics targeting the cancer epigenome can be grouped into two major categories: broad spectrum reprogrammers and narrowed spectrum reprogrammers [4]. An argument can be made for the potential effectiveness of both broad and targeted epigenetic therapies. Broad-spectrum reprogrammers include the inhibitors of DNA methyltransferase (DNMT), histone deacetylase (HDAC) and the bromodomain and extra-terminal motif proteins (BETs). These drugs cause genome-wide cancer-specific gene expression alterations. In contrast, narrowed spectrum epigenetic modifying agents targeting lysine-specific histone demethylase 1 (LSD1), enhancer of zeste homolog 2 (EZH2), DOT1-like histone lysine methyltransferase (DOT1L), to achieve precise inhibition of epigenetic regulatory proteins.
Broad spectrum reprogrammers
DNMT (DNA methyltransferase—“writer”) inhibitors
DNA methylation affects the transcription of genes without altering the DNA sequence. In eukaryotic DNA, cytosine is methylated and then converted into 5-methylcytosine by DNMTs [5]. Hypermethylation of specific regions, such as the CpG islands of tumor suppressor genes, plays an important role in carcinogenesis for many types of cancers [6,7,8]. There are 3 primary DNMTs—DNMT1, DNMT3A and DNMT3B [9,10,11]. DNMT1 is predominantly involved in maintaining the preexistent methylation pattern during DNA replication. DNMT3A and DNMT3B are involved in facilitating de novo DNA methylations at loci that were previously unmethylated [12]. Tumorigenesis often involves an interplay among all 3 DNMTs [13,14,15,16]. DNMT inhibitors act as cytidine analogs and induce loss of DNA methylation. There are two main classes of hypomethylating agents, the nucleoside analogs (such as 5-azacitidine that incorporates into DNA and RNA and 5-aza-2′-deoxycytidine, or decitabine, that incorporates into DNA) and the anti-sense DNA methyltransferase inhibitors (such as MG98) that do not require incorporation into DNA. The ability of azacitidine to be incorporated into DNA and RNA can lead to broad biological effects in resting and dividing cells [17]. DNMT inhibitors have shown to be particularly effective in targeting DNA methylation in leukemic cells [18, 19].
HDAC (histone deacetylase—“eraser”) inhibitors
Histone modification occurs via acetylation of lysine residues. Two families of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs), operate in an opposing manner. HATs acetylate lysines within the amino-terminal tails of histone proteins, resulting in relaxation of chromatin structure and facilitating gene activation. Conversely, HDACs remove acetyl groups from hyperacetylated histones and make the chromatin condensed and transcriptionally silent. There are four classes of HDAC enzymes based on their structures and functions: class I (HDAC 1–3 and 8), IIa (HDAC 4, 5, 7, 9), IIb (HDAC 6, 10), III (Sir-2 related—SIRT1-7) and IV (HDAC 11) [20, 21]. Class I HDAC proteins are mainly localized in the nucleus, whereas class II HDACs are expressed in a more tissue-restricted manner [22]. Sharing significant homology with both Class I and Class II HDACs, class IV HDAC does not possess a nuclear localization signal and its function is largely unknown [23]. HDACs are key elements in the regulation of gene expression, differentiation and development, and the maintenance of cellular homeostasis. HDAC inhibition causes global gene upregulation (potential oncosuppressors) and leads to arrest of tumor cell growth, apoptosis and anti-angiogenesis [24, 25]. In addition, HDAC facilitates the binding of elongation factors to acetylated promoters and enhancers for efficient elongation. Therefore, HDAC inhibitors block gene elongation and inhibit gene expression, especially in highly expressed genes (oncogenes) [26]. Many HDAC inhibitors are non-specific and can be used to inhibit multiple isoforms of HDACs.
BET (bromodomain and extra-terminal motif proteins—“reader”) inhibitors
BET proteins are known to recognize acetylated lysine in chromatin [27]. The BET family of proteins include BRD2, BRD3, BRD4, and the testes-specific BRDT [28, 29]. Bromodomains can specifically bind acetylated lysine residues of histone proteins, and are involved with histone modifications, chromatin remodeling and transcriptional activation via recruitment of other proteins [30, 31]. BRD2 and BRD3 facilitate the passage of RNA Pol II to elongate the DNA transcripts through hyperacetylated nucleosomes [32]. BRD4 enhances the recruitment of positive transcription elongation factor b (P-TEFb), leading to the release of Pol II from a pause in transcription elongation in the promoter-proximal region [33]. In particular, aberrant BRD4 expression contributes to carcinogenesis by mediating hyperacetylation of the chromatin associated with cell proliferation-promoting genes [34]. Suppression of BRD4 led to anti-leukemic effects in acute myeloid leukemia (AML) mouse models and revealed a potential epigenetic target for AML [35]. In addition, BRD4 and BET proteins also regulate enhancer (a short region of DNA that can be bound by transcription factors to enhance the transcription of a particular gene) function and, in particular, large clusters of enhancers (super-enhancers), which drive oncogene expression, such as BCL-2 and c-MYC [36, 37]. Interestingly, the pathogenic fusion product of NUT (nuclear protein in testis) with BRD4 or BRD3 (BRD4-NUT or BRD3-NUT) causes NUT midline carcinoma (NMC), which is a rare but poorly differentiated and highly aggressive cancer of the squamous cell lineage that arises in midline structures [38]. BET bromodomain blockade using small-molecule inhibitors leads to selective repression of the transcriptional network driven by c-MYC [39].
METTL3 (methyltransferase like-3—“writer”) inhibitors
In addition to the epigenetic modifications on either DNA or histones, methylation is also observed in eukaryotic RNAs, including messenger RNA (mRNA), microRNA (miRNA) and long non-coding RNA (lncRNA), etc. Methylation modification impacts RNA processing, nuclear export, translation initiation and degradation [40]. In particular, N6-methyladenosine (m6A) modification of mRNA is most abundant, which occurs in two consensus sequence motifs including G(m6A)C primarily and A(m6A)C to a lesser extent [41, 42]. m6A is installed by a multiprotein writer complex that consists of methyltransferase-like protein 3 (METTL3), methyltransferase-like protein 14 (METTL14) and other accessory subunits. m6A modification is reversible and it can be erased by ALKBH5 (alkB homolog 5) [43] and FTO (fat-mass and obesity associated protein) proteins (Fig. 1) [44]. In addition, METTL3 and METTL14 are also identified as key actors of adenosine methylation of miRNAs [45, 46], whereas FTO is recognized as a key actor of adenosine demethylation of miRNAs [47]. m6A reader proteins can specifically bind to m6A transcripts and regulate the metabolism of mRNA [48]. For example, YTHDF2 (YTH domain family 2) binds to m6A in mRNA and targets mRNA degradation, whereas YTHDF1, YTHDF3, and eukaryotic initiation factor 3 (eIF3) promote translation of mRNA transcripts [49]. METTL3 has been found to be upregulated with increased m6A levels in cancer compared with those in normal tissues, suggesting a potential oncogenic role in different cancer types including AML, renal cell carcinoma, non-small cell lung cancer (NSCLC) and gastric cancer [50,51,52,53]. The studies show that loss of either METTL14 or METTL3 in AML cell lines and primary leukemic blasts led to induction of differentiation [50, 54]. In addition, METTL3 has been associated with multiple cell signaling pathways, including tumorigenesis, proliferation, invasion, migration, cell cycle, differentiation and cell viability [55]. Currently, multiple METTL3 inhibitors are under investigation in both AML and solid tumors, with pending clinical trials in the near future [56].
Besides the role of METTL3 in m6A modification on mRNAs and miRNAs, recent study suggested that DNMT3A methylates miRNA at cytosine residues and inhibits the formation of miRNA/mRNA duplex, leading to the loss of their repressive function in gene expression [57]. Therefore, using demethylating agent to block miRNA methylation may broaden its therapeutic potentials.
Narrowed spectrum reprogrammers
LSD1 (histone demethylase—“eraser”) inhibitors
LSD1 (lysine-specific histone demethylase 1, also known as KDM1A) is the first discovered histone lysine demethylase with the ability to erase the mono-methyl and di-methyl chromatin marks on histone H3, predominantly at lysines 4 and 9 (H3K4 and H3K9) [58,59,60]. It can also demethylate non-histone proteins, including DNMT1 and TP53 [59]. Moreover, LSD1 is a multifunctional subunit of both repressive and activating histone-modifying complexes and can therefore act as both a transcriptional repressor or activator in a context-dependent manner [61]. LSD1 regulates the balance between self-renewal and differentiation of stem cells, and LSD1 inhibition in mixed lineage leukemia (MLL)-rearranged leukemia has been shown to downregulate expression of some leukemia associated genes and cause apoptosis and cell differentiation [62]. In addition, LSD1 is overexpressed in various solid tumors including prostate, breast, lung and colorectal cancers, and neuroblastoma [63,64,65,66,67]. Pharmacological inhibition of LSD1 leads to inhibition of proliferation, differentiation, invasion, and migration in vitro and in vivo [68]. Thus, LSD1 inhibitors might be promising potential therapeutic options in a variety of cancers. Recently, it has been demonstrated that the effects of LSD1 inhibitors are particularly robust for small cell lung cancer (SCLC) through promotion of differentiation of tumor-enriched stem-like cells [69].
EZH2 (histone methyltransferase—“writer”) inhibitors
Several families of histone methyltransferases (HMT) that catalyze the methylation of specific lysine residues in histones H3 and H4 have been identified [70]. Unlike other histone modifications, which simply specify active or repressed chromatin states, histone lysine methylations confer active or repressive transcription depending on their positions and methylation states [71]. EZH2 (enhancer of zeste homolog 2), a histone methyltransferase and a catalytic component of polycomb repressive complex 2 (PRC2), catalyzes tri-methylation of histone H3 at lysine 27 (H3K27me3) to promote transcription silencing [72, 73]. Through modulating critical gene expression, EZH2 promotes cell survival, proliferation, epithelial-to-mesenchymal transition (EMT), invasion, and drug resistance of cancer cells [74]. EZH2 is activated by mutations (gain-of-function) in lymphoma [75], and EZH2 overexpression is associated with aggressiveness and worse clinical outcome in several solid tumors, including prostate, breast, bladder, and endometrial cancers, and melanoma [76,77,78]. The use of an EZH2 inhibitor demonstrated selective killing effect in cell lines carrying EZH2 activating mutations [79]. Several studies also identified a PRC2-independent function of EZH2 in transcriptional activation, involving transcription of androgen receptor (AR), estrogen receptor (ER) and Wnt signaling [80,81,82,83]).
DOT1L (histone methyltransferase—“writer”) inhibitors
Disruptor of telomeric silencing 1 (DOT1) is a novel class of HMT that was first identified to dysregulate gene silencing near telomeres in yeast [84]. DOT1-like (DOT1L) is the only known methyltransferase that deposits mono-, di-, and trimethyl marks on histone H3 lysine 79 (H3K79) in mammals. It participates in the regulation of transcription, differentiation and proliferation of normal cells. DOT1L has been shown to be critical for transformation by MLL fusion proteins in AML [85, 86]. Preclinical models demonstrate that MLL-driven leukemia is particularly sensitive to inhibition of DOT1L activity, and DOT1L inhibitors have been shown to specifically reduce H3K79 methylation marks and expression of MLL-fusions target genes in leukemic cells [87]. In addition, a recent study demonstrated the role of DOT1L in breast cancers that do not harbor a MLL translocation. DOT1L plays an important role in the initiation and progression of breast cancer by targeting the gene expression of EMT-promoting factors, suggesting DOT1L to be a therapeutic target for aggressive breast cancer [88]. While the pre-clinical studies showed promising activity of DOT1L inhibitors, the phase I study of DOTlL inhibitor, pinometostat, in adult and pediatric patients with relapsed or refractory leukemia demonstrated limited clinical response [89, 90].
IDH (isocitrate dehydrogenase) inhibitors
Mutations in genes encoding enzymes of the tricarboxylic acid (TCA) cycle can disrupt cell metabolism and alter the epigenetic landscape. For example, IDH1/2 enzymes metabolize isocitrate to α-ketoglutarate (α-KG) in the TCA cycle. α-KG serves as a co-factor for α-KG-dependent dioxygenases, including the ten-eleven translocation (TET) family of DNA demethylases and Jumonji family of histone demethylases. TET family of DNA methylases act on methylated DNA sequences, convert 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which will ultimately remove methyl groups and ensure the correct DNA methylation in the cell [91]. IDH1/2 mutations are found in several cancer types, including AML, gliomas, chondrosarcoma and intrahepatic cholangiocarcinoma [92, 93]. IDH mutations (gain-of-function) result in further processing of α-KG to 2-hydroxyglutarate (2-HG). This leads to the production of “oncometabolite” 2-HG, which inhibits TET family of DNA demethylases and Jumonji family of histone demethylases [94] and promotes tumorigenesis [95]. Accumulation of 2-HG in leukemic cells leads to increased DNA and histone methylation and results in blocked cell differentiation [96, 97]. Several small molecule inhibitors of both IDH1 and IDH2 have demonstrated reduction of 2-HG levels and differentiation of leukemic cells that carry the specific IDH mutations [98,99,100]. These effects also correlate with global changes in DNA methylation/histone modification state, suggesting that the phenotypic effects are, to some extent, secondary to rewiring transcriptional programs in the leukemic cells [101].
The aforementioned RNA demethylases, FTO and ALKBH5 which demethylate m6A, are α-KG-dependent dioxygenases [102,103,104]. m6A destabilizes transcripts and controls expression of key transcription factors in hematopoietic stem cells (HSCs) and human embryonic stem cells (ESCs) [105]. 2-HG suppresses FTO activity in leukemia cells, leading to decreased expression of the lineage transcription factor CCAAT enhancer binding protein α (C/EBPα) that enforces normal HSC quiescence and myeloid differentiation [106]. Therefore, the inhibition of IDH may lead to the changes in metabolic activities in TCA cycle such as α-KG and 2-HG, coordinating the cell fate in HSCs and ESCs.
Epigenetic drugs for cancer treatment: approved or in clinical trials
Approved epigenetic therapies
To date, the FDA-approved epigenetic agents are mostly limited in treating hematologic malignancies. Two DNMT inhibitors are approved for the treatment of myelodysplastic syndrome (MDS)—azacitidine and decitabine. Clinical trials with azacitidine and its deoxy derivative, decitabine, demonstrated that 15% or more of the patients with AML or intermediate to high-risk MDS showed improvement in blood cell counts and survival [107, 108]. Several HDAC inhibitors are approved for the treatment of hematologic malignancies, including belinostat for peripheral T cell lymphoma (PTCL), panobinostat for multiple myeloma, vorinostat for cutaneous T cell lymphoma (CTCL) and romidepsin for both CTCL and PTCL. IDH inhibitors, enasidenib and ivosidenib, have been approved for relapsed or refractory AML with IDH mutations [109,110,111]. EZH2 inhibitor, Tazemetostat, has been approved for patients with relapsed or refractory follicular lymphoma (R/R FL) with EZH2 mutation and who have received at least 2 prior systemic therapies, and for adult patients with R/R FL who have no satisfactory alternative treatment options [112].
Clinical trials are ongoing in solid tumors with agents from multiple drug classes. In January 2020, tazemetostat has been granted accelerated approval by FDA in treating epithelioid sarcoma, for which we will discuss later in this article [113]. These FDA-approved agents are summarized in Table 1.
Monotherapies in solid tumors
Historically, the first generation DNMT inhibitors (azacytidine and decitabine) showed limited activity in solid tumor, in part due to their toxicity. Biomarker studies demonstrated evidence of DNA methylation changes associated with drug administration; however, the responses were short-lived and treatment resistance developed early [114,115,116,117]. A phase I study of decitabine was conducted in patients with stage IV lung cancer, esophageal cancer, and malignant pleural mesothelioma. No objective response was observed and severe toxicities occurred. Grade 4 neutropenia was observed in 43% (15 out of 35) of the patients and grade 3 hepatotoxicity were seen in two patients with extensive liver metastases [118].
The second-generation DNMT inhibitors, such as guadecitabine (SGI-110), have been undergoing investigation. Guadecitabine is a novel hypomethylating prodrug of decitabine with a prolonged half-life. This novel compound is an oligonucleotide consisting of decitabine linked through a phosphodiester bond to the endogenous nucleoside deoxyguanosine. The dinucleotide configuration provides protection from drug clearance [119]. Guadecitabine [119] has been demonstrated to be safe and well tolerated as a single agent, with evidence of promising activity in heavily pretreated MDS and AML patients [120]. A phase II trial of SGI-110 monotherapy in patients with HCC who progressed on sorafenib (NCT01752933) was completed. The single agent SGI-110 demonstrated disappointing PFS in this trial.
Similar to DNMT inhibitors, HDAC inhibitors have shown limited single agent activity, and responses have been rare in solid tumors [121, 122]. A phase II study of vorinostat in relapsed non-small cell lung cancer (NSCLC) showed no objective response in 14 evaluable patients, and severe toxicities were reported including neutropenia, lymphopenia, fatigue and pulmonary embolisms [123]. A phase III trial of vorinostat as second-line monotherapy in advanced mesothelioma was conducted in patients who had previously received chemotherapy, and it showed that single agent vorinostat did not improve overall survival (OS) compared with placebo [124]. Representative recent clinical trials of single agent DNMT inhibitors and HDAC inhibitors in solid tumors are summarized in Table 2.
To further explore the efficacy of epigenetic monotherapy, newer epigenetic agents have been investigated beyond HDAC and DNMT inhibitors, targeting more specific patient population with a narrowed spectrum epigenetic modulation. Among them, tazemetostat is the first FDA-approved epigenetic therapy in the solid tumor, epithelioid sarcoma [112]. ES is a rare soft tissue sarcoma that is characterized by the loss of expression in INI1/SNF5/SMARCB1. SMARCB1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1), a subunit of SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex, can repress EZH2 transcription [125]. The loss of INI1 function leads to elevated expression and recruitment of EZH2 to target genes, resulting in the upregulation of several oncogenic signaling pathways [126]. The accelerated approval of tazemetostat was based on the results of a single arm cohort in patients with metastatic or locally advanced ES who are not eligible for complete resection (NCT02601950). Nine out of sixty two patients with INI1-negative ES (15%) had partial response (PR) and six out of those nine patients (67%) had a duration of response lasting 6 months or longer. Tazemetostat was generally well tolerated [127] in the study.
In addition, early phase studies demonstrated BET inhibitors had clinical activities in patients with NMC. NMC is a rare and aggressive squamous cancer, which is commonly driven by the BRD4-NUT or BRD3-NUT fusion oncoprotein. A phase Ib study of birabresib (MK-8628/OTX015) was conducted in patients with NMC. Three out of ten patients (30%) with NMC had a PR with duration of response of 1.4 to 8.4 months [128]. In another phase I study of molibresib (GSK525762), out of nineteen NMC patients, four (21%) achieved either confirmed or unconfirmed PR and eight patients (42%) had stable disease as best response [129]. These results have demonstrated that targeting BRD4-NUT and BRD3-NUT with BET inhibitors resulted in strong antitumor activity in this rare patient population.
Another new epigenetic agent targeting a specific genetic defect in epigenetic pathways has been investigated. The phase III ClarIDHy trial (NCT02989857) evaluated the IDH1 inhibitor ivosidenib in 185 previously treated patients with IDH1-mutated advanced cholangiocarcinoma. Ivosidenib improved PFS from 1.4 months with placebo to 2.7 months (hazard ratio [HR] = 0.37; P < 0.001). Although the objective response rate was low (2.4%), clinical benefit was observed with stable disease (SD) in 50.8% of patients. Median OS was 10.8 months with ivosidenib versus 9.7 months with placebo (HR = 0.69; P = 0.06), including 57% of patients who crossed over from placebo group [130]. As a side note, the benefit of IDH1 inhibitors in patients with chondrosarcoma is controversial [131, 132], in part due to the different histological subtype with various disease aggressiveness and clinical outcome [133].
Summarized clinical trials investigating novel epigenetic drugs (single agent) in solid tumors are listed in Table 3.
Combination therapies in solid tumors
Due to the limited efficacy of epigenetic monotherapy as described previously, and the complexity of epigenetic modification in cancer, many trials are investigating combination therapies in solid tumors. Recent clinical trials include epigenetic modifier combinations as well as combinations of epigenetic agents with cytotoxic chemotherapy, hormonal therapies, and immune checkpoint inhibitors (ICIs).
Combination of DNMT inhibitors and HDAC inhibitors
Preclinical studies demonstrated that DNMT inhibitor enhances apoptosis in cancer cells induced by HDAC inhibitors, suggesting the potential synergism of DNMT in combination with HDAC inhibitors [134]. A phase I/II trial of azacitidine and entinostat in NSCLC yielded some promising results with durable responses [135]. This trial included heavily pre-treated patients who had received a median of three prior therapies. Clinical efficacy was observed with one complete response (CR) for 14 month duration, one PR for eight month duration, and ten patients with SD lasting at least 12 weeks. One of these patients had stable disease for 18 months and another for 14 months. The prolonged clinical benefit in certain patients in this trial prompted a correlative biomarker study to predict treatment response. The study collected and examined the promoter methylation status in circulating DNA from patient plasma collected before therapy (day 0) and after 1 cycle of therapy (day 29). Of these, ten out of 26 patients demonstrated a decrease in methylation during the first four weeks of treatment compared to their baseline. There was a higher response rate and improvement in overall survival in the patients with methylation changes (“methylation signature”–positive) compared to patients without methylation change (“methylation signature”–negative). The median OS and PFS were 10.42 months for the methylation signature-positive cohort versus 6.54 months for the methylation signature-negative (P = 0.035). This suggests a potential role of epigenetic therapy in NSCLC, and the important role of biomarkers to predict response and benefit in patients.
Epigenetic therapy with cytotoxic chemotherapy
Preclinical studies suggested that DNMT and HDAC inhibitors have the greatest efficacy when combined with chemotherapy in an attempt to re-sensitize cancers to the standard cytotoxic agents [136, 137]. Acquired resistance to the chemotherapy agents might be reversed when combined with DNMT and/or HDAC inhibitors, especially in ovarian cancers [138]. A phase I trial of low-dose decitabine combined with carboplatin was conducted in patients with recurrent platinum-resistant ovarian cancer. The low dose decitabine was tolerated and demonstrated biological activity in DNA hypomethylation. However, the clinical benefit was modest [139]. Another phase II randomized study compared guadecitabine in combination with carboplatin against second-line chemotherapy in patients with platinum-resistant ovarian cancer. It does not meet the primary endpoint and there is no difference in either median PFS or OS between the two groups [140, 141]. Similarly, in a phase I trial in patients with metastatic colorectal cancer who were exposed to irinotecan previously, guadecitabine in combination with irinotecan showed modest clinical activity with stable disease as the best response [142]. As a note, the challenge in epigenetic agents in combination with cytotoxic chemotherapies include the side effects of additive toxicities needing dose reduction of epigenetic agents. In addition, the chemotherapies cause G1/S cell cycle arrest, which may interfere with incorporation of hypomethylating agents into the DNA and RNA.
Epigenetic therapy with immune checkpoint inhibitors
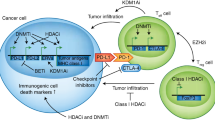
ICIs have recently changed the cancer treatment landscape in many types of cancers. The combination of epigenetic agents with ICIs is an area of investigation in a variety of solid tumors [143]. In the clinical trial involving 45 patients with advanced-stage NSCLC who were treated with azacitidine and entinostat, five patients who had disease progression during the trial were subsequently enrolled in trials of anti-PD-1 therapy [135]. Three of the five patients achieved an objective response and the other two had SD for 24 weeks before disease progression. This clinical observation has led to pre-clinical research to understand the mechanism of epigenetic therapies in modulating immune responses. Treatment of tumor cells with DNMT inhibitors can induce the transcription of endogenous retrovirus (ERVs), which are normally silenced in most somatic tissues [144]. The reactivation of ERVs result in the formation of cytoplasmic double-stranded RNAs [145, 146], the cognate ligand of the retinoic acid inducible gene I (RIG-I)-like receptors (RLR), including RIG-I and melanoma differentiation associated gene 5 (MDA5) [147]. Activation of the RLR family (innate immune sensors) initiates signaling cascades leading to the production of type I and III interferons, which elicit an antitumor immune response (virial mimicry) by activation of CD8+ T cells [148, 149]. Also, epigenetic therapy can lead to the re-expression of tumor antigens, such as cancer testis antigens (CTAs) and melanoma-associated antigen 1 (MAGE1), increasing immunogenicity [150,151,152]. Therefore, both pre-clinical and clinical studies suggests that these epigenetic therapies might augment antitumor immune response through various mechanisms, enhancing tumor antigen expression and infiltration of cytotoxic T cells, and reversing T cell exhaustion with a concurrent increase in the abundance of effector and/or memory T cells, among others [153]. These observations are being translated into clinical trials that focus on the combination of ICIs with epigenetic drugs in a variety of solid tumors.
A phase I/Ib trial of pembrolizumab plus oral vorinostat (HDAC inhibitor) has been completed in patients with advanced/metastatic NSCLC [154]. Thirty-three patients were treated, including thirteen in phase I and twenty in phase Ib. In phase I, both ICI-naïve and ICI-pretreated patients were enrolled to determine dose-limiting toxicities (DLTs). No DLTs were observed, and the recommended phase II dose was pembrolizumab 200 mg and vorinostat 400 mg/day. The most common adverse events of any grade included fatigue (33%) and nausea/vomiting (27%). Among those 6 ICI-naïve patients, there was 1 case (16.7%) of confirmed PR, 4 cases (66.7%) of SD, and 1 case (16.7%) of PD. Of 24 ICI-pretreated patients evaluable for response, there were 3 cases with (13%) PR (1 confirmed), 11 cases with (46%) SD and 10 cases (42%) with progressive disease (PD). The results suggested the combined therapy of pembrolizumab and vorinostat is feasible with a manageable safety profile and active in both ICI-naïve and -exposed NSCLC patients. The presence of CD8+ T-cell in tumor stroma in pretreatment samples, not CD8+ T-cell in tumor bed, was associated with treatment benefit. In addition, on-treatment biopsies showed the increase in CD8+ T cells in the stroma was correlated with clinical benefit (with SD or PR for a period of ≥ 24 weeks). It would be crucial to investigate whether the combination is better than ICI alone in ICI-naïve patients in the front line setting and/or if the combination is superior to the standard of care in ICI-exposed patients in the later line treatment setting. An ongoing randomized phase 2 trial is examining pembrolizumab +/− vorinostat in ICI-naive advanced/metastatic NSCLC patients (NCT02638090).
Similarly, a phase II study is investigating azacitidine and entinostat with concurrent nivolumab in patients with metastatic NSCLC, in both ICI-naïve and ICI-resistant patient populations (NCT01928576) and a phase I study is investigating pembrolizumab in combination with guadecitabine and mocetinostat for patients with advanced lung cancer who progressed on prior ICIs (NCT03220477). These on-going trials include correlative studies to evaluate induced viral mimicry, interferon induction, and T cell function phenotypes [153].
The newer epigenetic agents in combination with ICIs are also under investigation. A phase I/II trial is evaluating a BET inhibitor, INCB057643, in combination with pembrolizumab and epacadostat (indoleamine 2, 3-dioxygenase or IDO-1 inhibitor) in patients with advanced or metastatic solid tumors (NCT02959437). Additionally, trials of EZH2 inhibitors in combination with ipilimumab (CTLA-4 inhibitor) or pembrolizumab are recruiting the patients with advanced solid tumors (NCT03525795 and NCT03854474).
Epigenetic therapy with other anticancer therapies
New approaches combining epigenetic agents with other anticancer therapies, including hormonal therapy, have been explored as an approach to overcome treatment resistance. In the phase II study ENCORE301, entinostat was added to exemestane (steroidal aromatase inhibitor [AI]) in patients with hormone receptor (HR)-positive advanced breast cancer with disease progression after prior non-steroidal AI. The study demonstrated a significant improvement in PFS (HR = 0.73; p = 0.06) and also in OS (HR = 0.59; p = 0.036). The combination was well tolerated, with neutropenia (13%) and fatigue (11%) being the most frequent grade 3 or 4 toxicities in entinostat-treated patients [155]. Therefore, entinostat, when added to exemestane, was designated by the FDA as breakthrough therapy for postmenopausal women with HR-positive advanced breast cancer whose disease has progressed after nonsteroidal AI therapy. Based on the ENCORE301 study, a phase III trial (E2112) is ongoing to investigate entinostat versus placebo in combination with exemestane in patients with locally advanced or metastatic breast cancer who have experienced disease progression after a non-steroidal AI [156]
Everolimus, a sirolimus (formerly called rapamycin) derivative, inhibits phosphatidylinositol 3-kinase (PI3K)/Akt/(158)mammalian target of rapamycin (mTOR) signaling pathway, which is one of the mechanisms of endocrine resistance in HR-positive breast cancer [157, 158]. In preclinical studies, the use of everolimus in combination with aromatase inhibitors results in synergistic inhibition of the proliferation and induction of apoptosis [159]. The BOLERO-2 trial showed that everolimus in combination with exemestane improved PFS compared to exemestane alone in post-menopausal women with advanced HR+/Her2-negative breast cancer [160]. However, recent data suggested that the combination of exemestane and everolimus did not yield a durable clinical response, indicating a need for alternative combinations and therapeutic strategies [161]. The pre-clinical studies showed that resistance to everolimus was mediated by overexpression of MYC in ER-positive cancers, which can be reversed by BET inhibitors [162]. Also, a combination of BET inhibitor with fulvestrant (ER degrader) showed long-lasting antitumor effect in a tamoxifen (selective ER modulator)-resistant breast cancer xenograft mouse model [163].
Similarly, the combination of BET inhibitors with AR antagonists is able to subvert resistance in castrate-resistant prostate cancer (CRPC) in preclinical experiments [164]. Other studies combining BET and PARP inhibition show mitotic catastrophe (cell death related to premature entry of cells into mitosis) with induction of apoptosis, causing synergistic effect in suppressing BRCA1/2 wild-type ovarian cancer. This study also suggests that BET inhibitors re-sensitize PARP-inhibitor-resistant BRCA mutant epithelial ovarian cancer cells to PARP inhibition [165]. DNMT inhibitors create a “BRCAness” phenotype through downregulating expression of key homologous recombination and nonhomologous end-joining (NHEJ) genes, and promote synergism with PARP inhibitors in the setting of BRCA-proficient NSCLC in animal models. These pre-clinical data support the expansion of therapeutic studies of PARP inhibitors and various epigenetic agents in patients with BRCA-proficient cancer [166].
There are also ongoing clinical trials with BET inhibitors in combination with PARP inhibitors, ER antagonists, and AR antagonists. A phase I trial is accruing patients to investigate AZD5153 in combination with olaparib for platinum-resistant/refractory ovarian cancer. Other accruing studies include a phase II trial of ZEN003694 in combination with talazoparib in TNBC (NCT03901469); a phase I/II trial to test GSK525762 in combination with fulvestrant in advanced HR-positive breast cancer (NCT02964507); and a phase Ib study combining GSK525762 with abiraterone or enzalutamide in advanced CRPC (NCT03150056). In addition, several early phase trials are investigating EZH2 inhibitors in combination with enzalutamide or abiraterone in metastatic CRPC, given the synergistic effect of EZH2 inhibitors in combination with AR antagonists.
Ongoing clinical trials of combination therapies of epigenetic drugs with chemotherapy or other agents including ICIs in solid tumors are listed in Table 4.
Conclusions
The development of epigenetic therapeutics has promise for cancer treatment, particularly with advancements in hematologic malignancies. In solid tumors, only one epigenetic agent (EZH2 inhibitor, tazemetostat) has been approved (ES). It is not fully understood why solid tumors are not as sensitive to epigenetic agents, even though there is profound aberrant epigenetic alterations in solid tumors. There may be a critical difference in cellular differentiation and epigenetic plasticity between solid tumors and hematological malignancies. Solid tumors arise from a more terminally differentiated state, which may be intrinsically more resistant to epigenetic reprogramming. In contrast, hematopoietic lineages are precisely controlled by epigenetic modulation. It is understandable that epigenetic agents demonstrated robust clinical activity in hematological malignancies in which cell differentiation is a key biological feature. The alternative explanation could be that altered epigenetic modulation may occur early in oncogenesis, however, it is not the “driver” event that controls the tumor cell proliferation and survival [167]. In the era of precision oncology, the broad impact of epigenetic treatment is both promising in “reprograming” solid tumor epigenetic dysfunction, as well as challenging in targeting particular epigenetic driving events. In recent years, the further development of next generation of broad spectrum agents and the emerging narrow spectrum agents as potential targeted epigenetic therapy have provided the new opportunities for solid tumor therapy. The approval of an epigenetic agent (EZH2 inhibitor, tazemetostat) in treatment of a rare soft tissue malignancy, epithelioid sarcoma, is a solid step towards the future breakthrough in the mechanism based solid tumor epigenetic treatment.
Various HDAC and DNMT inhibitors have been tested for treatment of both hematologic malignancies and solid tumors. Primary and secondary resistance to these therapies are common [168, 169]. No clear clinical benefits have been observed as yet in solid tumors. The limited antitumor activity with DNMT and HDAC inhibitors as monotherapy in solid tumors may also be related to either the short half-lives of the S phase-specific drugs with low incorporation into DNA [115] or due to a lack of specificity. Combination therapies with dual DNMT and HDAC inhibitors are explored in clinical trials; the therapeutic rationale is that densely methylated DNA is usually accompanied by deacetylated histone (transcriptionally repressive) [170]. However, most of the dual-agent epigenetic therapy trials did not result in an obvious clinical benefit, except the observation of durable responses in select NSCLC patients [135].
Potential novel therapies are being investigated to target new epigenetic modulation, such as IDH mutation inhibition and LSD1 inhibition, in both hematologic and solid malignancies. Many of these agents are targeting specific genetic defects in epigenetic pathways. Ivosidenib showed improved PFS in patients with cholangiocarcinoma harboring IDH1 mutation [130]. Pre-clinical studies suggest targeted epigenetic therapy may be effective in specific patient subsets, such as LSD1 inhibitors in the treatment for SCLC [69]. Early phase studies demonstrated BET inhibitors had activities in NMC, which is driven by BET fusion proteins. Most recently, METTL3 inhibitors and other agents targeting RNA epigenetics are emerging as potential cancer therapies with pending clinical trials.
The exciting finding that epigenetic agents are able to modulate tumor microenvironment has been a focus of epigenetic research. The combination of these “reprogramming” effects with other approved or novel therapies are being extensively explored. One of the current focuses is the combined epigenetic and immune therapy. It may be speculated that epigenetic agents have a significant “reprogramming” activity in immune cell components in addition to cancer cell component. There are many ongoing clinical trials evaluating the combination of the epigenetic agents with ICI in solid tumors. DNMT, HDAC, and other epigenetic inhibitors may enhance the response to and/or reverse the resistance to ICIs, if these agents can modulate key components of the tumor microenvironment including tumor cells, stromal cells, and innate and/or adaptive immune cells.
Beyond the scope of the current review, there are also important implications of epigenetic biomarkers in cancer screening, diagnosis, prognosis, and prediction to treatment. The development in the epigenetic biomarkers field are addressed in other reviews, including this one by Berdasco et al. [171].
In summary, epigenetic drugs represent “genomic medicines” that do not require existing DNA mutations. Given the wide diversity of solid tumors, epigenetic therapy is attractive because of the potential to target and modify the cancer genome functions. It is likely that cancer cells exploit epigenetic modulation to activate cellular pathways in cancer cell survival, including drug resistance and immune surveillance. Thus, epigenetic agents may have great therapeutic potential in the future under the right contexts. It will be essential to continue fundamental research to better identify the underlying mechanism and to translate these findings into clinical trial of newer epigenetic agents and optimize combinatorial approaches with exploration of predictive biomarkers in solid tumors.
Availability of data and materials
Not applicable.
Abbreviations
- 2-HG:
-
2-Hydroxyglutarate
- 5caC:
-
5-Carboxylcytosine
- 5fC:
-
5-Formylcytosine
- 5hmC:
-
5-Hydroxymethylcytosine
- 5mC:
-
5-Methylcytosine
- ALKBH5:
-
AlkB homolog 5
- AML:
-
Acute myeloid leukemia
- AR:
-
Androgen receptor
- BCL-2:
-
B cell lymphoma 2
- BET:
-
Bromodomain and extra-terminal motif proteins
- BRD:
-
Bromodomain
- c-MYC:
-
Cellular myelocytomatosis gene
- CR:
-
Complete response
- CTCL:
-
Cutaneous T cell lymphoma
- DNA:
-
Deoxyribonucleic acid
- DNMT:
-
DNA methyltransferase
- DOT1L:
-
DOT1-like histone lysine methyltransferase
- eIF3:
-
Eukaryotic initiation factor 3
- ER:
-
Estrogen receptor
- ES:
-
Epithelioid sarcoma
- EZH2:
-
Enhancer of zeste homolog 2
- FDA:
-
U.S. Food and Drug Administration
- FTO:
-
Fat-mass and obesity associated protein
- GSK525762:
-
Molibresib
- H2A:
-
Histone 2A
- H2B:
-
Histone 2B
- H3B:
-
Histone 3B
- H4:
-
Histone 4
- HAT:
-
Histone acetyltransferases
- HDAC:
-
Histone deacetylase
- HMT:
-
Histone methyltransferases
- HR:
-
Hazard ratio
- ICI:
-
Immune checkpoint inhibitor
- IDH:
-
Isocitrate dehydrogenase
- LSD1:
-
Lysine-specific histone demethylase 1
- MDS:
-
Myelodysplastic syndrome
- METTL:
-
Methyltransferase-like protein
- MK-8628/OTX015:
-
Birabresib
- MLL:
-
Mixed-lineage lymphoma
- NMC:
-
NUT midline carcinoma
- NSCLC:
-
Non-small cell lung cancer
- NUT:
-
Nuclear protein in testis
- OS:
-
Overall survival
- PR:
-
Partial response
- PRC2:
-
Polycomb repressive complex 2
- PTCL:
-
Peripheral T cell lymphoma
- P-TEFb:
-
Positive transcription elongation factor b
- R/R FL:
-
Relapsed/refractory follicular lymphoma
- RNA:
-
Ribonucleic acid
- SCLC:
-
Small cell lung cancer
- SD:
-
Stable disease
- SGI-110:
-
Guadecitabine
- SIRT:
-
Sir-2 related
- SMARCB1/INI:
-
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1
- SWI/SNF:
-
Switch/Sucrose non-fermentable
- TCA:
-
Tricarboxylic acid
- TET:
-
Ten-eleven translocation
- TP53:
-
Tumor protein 53
- YTHDF:
-
YTH domain family
- α-KG:
-
α-Ketoglutarate
References
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400.
Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–49.
Verschraegen CF, Stein G. Methylation, Methionine and Metaphors. In: Srivastava R, Maksymowicz W, Lopaczynski W, editors. Lost In Translation: Barriers To Incentives For Translational Research In Medical Sciences. 1st ed: World Scientific; 2014. p. 21–43.
Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–41.
Cheng X, Blumenthal RM. Mammalian DNA methyltransferases: a structural perspective. Structure. 2008;16(3):341–50.
McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther. 2015;150:1–22.
Gigek CO, Chen ES, Calcagno DQ, Wisnieski F, Burbano RR, Smith MA. Epigenetic mechanisms in gastric cancer. Epigenomics. 2012;4(3):279–94.
Saghafinia S, Mina M, Riggi N, Hanahan D, Ciriello G. Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep. 2018;25(4):1066–80 e8.
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–54.
Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11(10):726–34.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92.
Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236(1):87–95.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19(3):219–20.
Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404(6781):1003–7.
Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416(6880):552–6.
Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE. 2010;5(2):e9001.
Wilson VL, Jones PA, Momparler RL. Inhibition of DNA methylation in L1210 leukemic cells by 5-aza-2’-deoxycytidine as a possible mechanism of chemotherapeutic action. Cancer Res. 1983;43(8):3493–6.
Momparler RL, Bouchard J, Onetto N, Rivard GE. 5-aza-2’-deoxycytidine therapy in patients with acute leukemia inhibits DNA methylation. Leuk Res. 1984;8(2):181–5.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–91.
Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6(6):579–89.
Morris MJ, Monteggia LM. Unique functional roles for class I and class II histone deacetylases in central nervous system development and function. Int J Dev Neurosci. 2013;31(6):370–81.
Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1(1):19–25.
Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19(9):1049–66.
Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60(18):5165–70.
Greer CB, Tanaka Y, Kim YJ, Xie P, Zhang MQ, Park I-H, et al. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 2015;13(7):1444–55.
Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513(1):124–8.
Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282(18):13141–5.
Pivot-Pajot C, Caron C, Govin J, Vion A, Rousseaux S, Khochbin S. Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol Cell Biol. 2003;23(15):5354–65.
Winston F, Allis CD. The bromodomain: a chromatin-targeting module? Nat Struct Biol. 1999;6(7):601–4.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5.
LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol Cell. 2008;30(1):51–60.
Kanno T, Kanno Y, LeRoy G, Campos E, Sun HW, Brooks SR, et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol. 2014;21(12):1047–57.
Taniguchi Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int J Mol Sci. 2016;17(11):66.
Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–8.
Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34.
Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28(2):311–20.
French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003;63(2):304–7.
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17.
Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol. 2018;11(1):48.
Wei C-M, Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16(8):1672–6.
Wei CM, Gershowitz A, Moss B. 5’-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry. 1976;15(2):397–401.
Zheng G, Dahl John A, Niu Y, Fedorcsak P, Huang C-M, Li Charles J, et al. ALKBH5 Is a Mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6)-methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–43.
Berulava T, Rahmann S, Rademacher K, Klein-Hitpass L, Horsthemke B. N6-adenosine methylation in MiRNAs. PLoS ONE. 2015;10(2):e0118438.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361(6409):1346–9.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–200.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue Q, et al. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. 2017;482(4):582–9.
Li X, Tang J, Huang W, Wang F, Li P, Qin C, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8(56):66.
Liu T, Yang S, Sui J, Xu S-Y, Cheng Y, Shen B, et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J Cell Physiol. 2020;235(1):548–62.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. 2018;22(2):191-205.e9.
Zheng W, Dong X, Zhao Y, Wang S, Jiang H, Zhang M, et al. Multiple functions and mechanisms underlying the role of METTL3 in human cancers. Front Oncol. 2019;9:66.
Cully M. Chemical inhibitors make their RNA epigenetic mark. Nat Rev Drug Discov. 2019;18(12):892–4.
Cheray M, Etcheverry A, Jacques C, Pacaud R, Bougras-Cartron G, Aubry M, et al. Cytosine methylation of mature microRNAs inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol Cancer. 2020;19(1):36.
Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8(11):829–33.
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53.
Hosseini A, Minucci S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics. 2017;9(8):1123–42.
Rudolph T, Beuch S, Reuter G. Lysine-specific histone demethylase LSD1 and the dynamic control of chromatin. Biol Chem. 2013;394(8):1019–28.
Feng Z, Yao Y, Zhou C, Chen F, Wu F, Wei L, et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol. 2016;9:24.
Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31(3):512–20.
Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, et al. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer. 2013;109(4):994–1003.
Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66(23):11341–7.
Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, et al. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE. 2012;7(4):e35065.
Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69(5):2065–71.
Zheng YC, Yu B, Chen ZS, Liu Y, Liu HM. TCPs: privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics. 2016;8(5):651–66.
Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28(1):57–69.
Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12(2):198–209.
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48(4):491–507.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9.
Di Croce L, Helin K. Transcriptional regulation by polycomb group proteins. Nat Struct Mol Biol. 2013;20(10):1147–55.
Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res. 2018;6:10.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–9.
Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22(20):5323–35.
Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24(2):268–73.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12.
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338(6113):1465–9.
Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27(14):5105–19.
Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78–83.
Xiao L, Tien JC, Vo J, Tan M, Parolia A, Zhang Y, et al. Epigenetic reprogramming with antisense oligonucleotides enhances the effectiveness of androgen receptor inhibition in castration-resistant prostate cancer. Cancer Res. 2018;78(20):5731–40.
Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, et al. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150(2):613–32.
Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, et al. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 2010;70(24):10234–42.
Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117(18):4759–68.
Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78.
Lee JY, Kong G. DOT1L: a new therapeutic target for aggressive breast cancer. Oncotarget. 2015;6(31):30451–2.
Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131(24):2661–9.
Shukla N, Wetmore C, O’Brien MM, Silverman LB, Brown P, Cooper TM, et al. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with relapsed or refractory MLL-r acute leukemia. Blood. 2016;128(22):2780.
Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517–34.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–66.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73.
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8.
Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–5.
Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–6.
Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122(16):2877–87.
Gallipoli P, Huntly BJP. Novel epigenetic therapies in hematological malignancies: current status and beyond. Semin Cancer Biol. 2018;51:198–210.
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318(5855):1469–72.
Xu C, Liu K, Tempel W, Demetriades M, Aik W, Schofield CJ, et al. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J Biol Chem. 2014;289(25):17299–311.
Gerken T, Girard CA, Tung Y-CL, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated fto gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318(5855):1469–72.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22(2):191–205e9.
Baksh SC, Finley LWS. Metabolic coordination of cell fate by α-ketoglutarate-dependent dioxygenases. Trends Cell Biol. 2021;31(1):24–36.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32.
Lubbert M, Suciu S, Hagemeijer A, Ruter B, Platzbecker U, Giagounidis A, et al. Decitabine improves progression-free survival in older high-risk MDS patients with multiple autosomal monosomies: results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group. Ann Hematol. 2016;95(2):191–9.
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–31.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–98.
Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2020;135(7):463–71.
FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer [press release]. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-option-specifically-patients-epithelioid-sarcoma-rare-soft-tissue. Accessed 23 January 2020.
FDA granted accelerated approval to tazemetostat for follicular lymphoma [press release]. https://www.fda.gov/drugs/fda-granted-accelerated-approval-tazemetostat-follicular-lymphoma#:~:text=On%20June%2018%2C%202020%2C%20the,approved%20test%20and%20who%20have. Accessed 18 June 2020.
Weiss AJ, Metter GE, Nealon TF, Keanan JP, Ramirez G, Swaiminathan A, et al. Phase II study of 5-azacytidine in solid tumors. Cancer Treat Rep. 1977;61(1):55–8.
Stewart DJ, Issa JP, Kurzrock R, Nunez MI, Jelinek J, Hong D, et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009;15(11):3881–8.
Samlowski WE, Leachman SA, Wade M, Cassidy P, Porter-Gill P, Busby L, et al. Evaluation of a 7-day continuous intravenous infusion of decitabine: inhibition of promoter-specific and global genomic DNA methylation. J Clin Oncol. 2005;23(17):3897–905.
Aparicio A, Eads CA, Leong LA, Laird PW, Newman EM, Synold TW, et al. Phase I trial of continuous infusion 5-aza-2’-deoxycytidine. Cancer Chemother Pharmacol. 2003;51(3):231–9.
Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, et al. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12(19):5777–85.
Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR. SGI-110: DNA methyltransferase inhibitor oncolytic. Drugs Future. 2013;38(8):535–43.
Issa J-PJ, Roboz G, Rizzieri D, Jabbour E, Stock W, O'Connell C, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. The Lancet Oncology. 2015;16(9):1099–110.
Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009;280(2):192–200.
Qiu T, Zhou L, Zhu W, Wang T, Wang J, Shu Y, et al. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013;9(2):255–69.
Traynor AM, Dubey S, Eickhoff JC, Kolesar JM, Schell K, Huie MS, et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network Phase II Study. J Thorac Oncol. 2009;4(4):522–6.
Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D, et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015;16(4):447–56.
Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21(12):1491–6.
Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014;30(8):356–63.
Stacchiotti S, Schoffski P, Jones R, Agulnik M, Villalobos VM, Jahan TM, et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES) (NCT02601950). J Clin Oncol. 2019;37(15_suppl):11003.
Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol. 2018;36(30):3007–14.
Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 Study of Molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectr. 2019;4(2):66.
Abou-Alfa GK, Macarulla Mercade T, Javle M, Kelley RK, Lubner S, Adeva J, et al. LBA10_PR - ClarIDHy: A global, phase III, randomized, double-blind study of ivosidenib (IVO) vs placebo in patients with advanced cholangiocarcinoma (CC) with an isocitrate dehydrogenase 1 (IDH1) mutation. Ann Oncol. 2019;30:v872–3.
Cleven AHG, Suijker J, Agrogiannis G, Briaire-de Bruijn IH, Frizzell N, Hoekstra AS, et al. IDH1 or -2 mutations do not predict outcome and do not cause loss of 5-hydroxymethylcytosine or altered histone modifications in central chondrosarcomas. Clin Sarcoma Res. 2017;7:8.
Lugowska I, Teterycz P, Mikula M, Kulecka M, Kluska A, Balabas A, et al. IDH1/2 mutations predict shorter survival in chondrosarcoma. J Cancer. 2018;9(6):998–1005.
Cojocaru E, Wilding C, Engelman B, Huang P, Jones RL. Is the IDH mutation a good target for chondrosarcoma treatment? Curr Mol Biol Rep. 2020;6(1):1–9.
Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2001;61(4):1327–33.
Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607.
Benson EA, Skaar TC, Liu Y, Nephew KP, Matei D. Carboplatin with decitabine therapy, in recurrent platinum resistant ovarian cancer, alters circulating miRNAs concentrations: a pilot study. PLoS ONE. 2015;10(10):e0141279.
Fang F, Zuo Q, Pilrose J, Wang Y, Shen C, Li M, et al. Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget. 2014;5(11):3579–89.
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80.
Fang F, Balch C, Schilder J, Breen T, Zhang S, Shen C, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116(17):4043–53.
Oza AM, Matulonis UA, Alvarez Secord A, Nemunaitis J, Roman LD, Blagden SP, et al. A randomized phase II trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant. Recurr Ovarian Cancer Clin Cancer Res. 2020;26(5):1009–16.
Oza AM, Matulonis UA, Alvarez Secord A, Nemunaitis J, Roman LD, Blagden SP, et al. A randomized phase 2 trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer. Clin Cancer Res. 2019:clincanres.1638.2019.
Lee V, Wang J, Zahurak M, Gootjes E, Verheul HM, Parkinson R, et al. A phase I trial of a guadecitabine (SGI-110) and irinotecan in metastatic colorectal cancer patients previously exposed to irinotecan. Clin Cancer Res. 2018;24(24):6160–7.
Cho JH, Oezkan F, Koenig M, Otterson GA, Herman JG, He K. Epigenetic therapeutics and their impact in immunotherapy of lung cancer. Curr Pharmacol Rep. 2017;3(6):360–73.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–86.
Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–73.
Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13(3):214–22.
Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015;6:29.
Karpf AR. A potential role for epigenetic modulatory drugs in the enhancement of cancer/germ-line antigen vaccine efficacy. Epigenetics. 2006;1(3):116–20.
Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2’-deoxycytidine. Cancer Res. 1994;54(7):1766–71.
Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002;21(35):5496–503.
Karpf AR, Peterson PW, Rawlins JT, Dalley BK, Yang Q, Albertsen H, et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci USA. 1999;96(24):14007–12.
Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17(2):75–90.
Gray JE, Saltos AN, Tanvetyanon T, Haura EB, Creelan BC, Antonia SJ, et al. Phase 1/1b study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin Cancer Res. 2019:clincanres.1305.2019.
Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31(17):2128–35.
Yeruva SLH, Zhao F, Miller KD, Tevaarwerk AJ, Wagner LI, Gray RJ, et al. E2112: randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. NPJ Breast Cancer. 2018;4(1):1.
Burstein HJ. Novel agents and future directions for refractory breast cancer. Semin Oncol. 2011;38:S17–24.
Johnston SRD. Clinical efforts to combine endocrine agents with targeted therapies against epidermal growth factor receptor/human epidermal growth factor receptor 2 and Mammalian target of rapamycin in breast cancer. Clin Cancer Res. 2006;12(3):1061s-s1068.
Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O’Reilly T, Evans DB, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005;11(14):5319–28.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2011;366(6):520–9.
Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2†. Ann Oncol. 2014;25(12):2357–62.
Bihani T, Ezell SA, Ladd B, Grosskurth SE, Mazzola AM, Pietras M, et al. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget. 2015;6(4):2407–20.
Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24(7):809–19.
Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara-Wilke J, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res. 2016;14(4):324–31.
Karakashev S, Zhu H, Yokoyama Y, Zhao B, Fatkhutdinov N, Kossenkov AV, et al. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017;21(12):3398–405.
Abbotts R, Topper MJ, Biondi C, Fontaine D, Goswami R, Stojanovic L, et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc Natl Acad Sci USA. 2019;116(45):22609–18.
Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019;25(3):403–18.
Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS ONE. 2011;6(8):e23372.
Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol. 2011;29(24):3322–7.
Eden S, Hashimshony T, Keshet I, Cedar H, Thorne AW. DNA methylation models histone acetylation. Nature. 1998;394(6696):842.
Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109–27.
Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and Leukemia Group B. J Clin Oncol. 2002;20(10):2429–40.
Vigil CE, Martin-Santos T, Garcia-Manero G. Safety and efficacy of azacitidine in myelodysplastic syndromes. Drug Des Devel Ther. 2010;4:221–9.
Khan C, Pathe N, Fazal S, Lister J, Rossetti JM. Azacitidine in the management of patients with myelodysplastic syndromes. Ther Adv Hematol. 2012;3(6):355–73.
Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–803.
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2006;109(1):31–9.
Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–7.
Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117(22):5827–34.
O’Connor OA, Horwitz S, Masszi T, Hoof AV, Brown P, Doorduijn J, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the Pivotal Phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492–9.
San-Miguel JF, Hungria VTM, Yoon S-S, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15(11):1195–206.
Mesia R, Bossi P, Hansen AR, Hsieh CY, Licitra LF, Tan EH, et al. Phase II study of CC-486 (oral azacitidine) in previously treated patients with locally advanced or metastatic nasopharyngeal carcinoma. Eur J Cancer. 2019;123:138–45.
Medicine NLo. SGI-110 in the Treatment of Advanced Hepatocellular Carcinoma (HCC) 2012 [updated January 18, 2020. NCT01752933]. https://clinicaltrials.gov/ct2/show/NCT01752933.
Grivas P, Mortazavi A, Picus J, Hahn NM, Milowsky MI, Hart LL, et al. Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes. Cancer. 2019;125(4):533–40.
Ibrahim N, Buchbinder EI, Granter SR, Rodig SJ, Giobbie-Hurder A, Becerra C, et al. A phase I trial of panobinostat (LBH589) in patients with metastatic melanoma. Cancer Med. 2016;5(11):3041–50.
Nilubol N, Merkel R, Yang L, Patel D, Reynolds JC, Sadowski SM, et al. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin Endocrinol. 2017;86(1):128–33.
Goncalves PH, Heilbrun LK, Barrett MT, Kummar S, Hansen AR, Siu LL, et al. A phase 2 study of vorinostat in locally advanced, recurrent, or metastatic adenoid cystic carcinoma. Oncotarget. 2017;8(20):32918–29.
Fan B, Mellinghoff IK, Wen PY, Lowery MA, Goyal L, Tap WD, et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Invest New Drugs. 2020;38(2):433–44.
Mellinghoff IK, Cloughesy TF, Wen PY, Taylor JW, Maher EA, Arrillaga I, et al. A phase I, open label, perioperative study of AG-120 and AG-881 in recurrent IDH1 mutant, low-grade glioma: Results from cohort 1. J Clin Oncol. 2019;37(15_supppl):2003.
Wang JS-Z, Vita SD, Karlix JL, Cook C, Littlewood GM, Hattersley MM, et al. First-in-human study of AZD5153, a small molecule inhibitor of bromodomain protein 4 (BRD4), in patients (pts) with relapsed/refractory (RR) malignant solid tumor and lymphoma: preliminary data. J Clin Oncol. 2019;37(15_suppl):3085.
Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 study of molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectrum. 2019.
Zauderer MG, Szlosarek P, Moulec SL, Popat S, Taylor P, Planchard D, et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. J Clin Oncol. 2018;36(15_suppl):8515.
Connolly RM, Li H, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC, et al. Combination epigenetic therapy in advanced breast cancer with 5-azacitidine and entinostat: a phase II National Cancer Institute/stand up to cancer study. Clin Cancer Res. 2017;23(11):2691–701.
Gaillard SL, Zahurak M, Sharma A, Durham JN, Reiss KA, Sartorius-Mergenthaler S, et al. A phase 1 trial of the oral DNA methyltransferase inhibitor CC-486 and the histone deacetylase inhibitor romidepsin in advanced solid tumors. Cancer. 2019;125(16):2837–45.
Hsieh WS, Tan EH, Lim W-T, Soo RA, Chan ATC, Hui EP, et al. A phase IB trial of 5-azacitidine (5AC) and suberoylanilide hydroxamic acid (SAHA) in patients with metastatic or locally recurrent nasopharyngeal carcinoma (NPC) and NK-T cell lymphoma. J Clin Oncol. 2013;31(15_suppl):17017.
Overman MJ, Morris V, Moinova H, Manyam G, Ensor J, Lee MS, et al. Phase I/II study of azacitidine and capecitabine/oxaliplatin (CAPOX) in refractory CIMP-high metastatic colorectal cancer: evaluation of circulating methylated vimentin. Oncotarget. 2016;7(41):67495–506.
Medicine NLo. A Phase I/II Clinical Trial of Vidaza With Abraxane in Patients With Advanced/Metastatic Solid Tumors and Breast Cancer (VA) 2008 [updated July 26, 2017. NCT00748553]. https://www.clinicaltrials.gov/ct2/show/NCT00748553.
Morgensztern D, Cobo M, Ponce Aix S, Postmus PE, Lewanski CR, Bennouna J, et al. ABOUND2.L+: A randomized phase 2 study of nanoparticle albumin-bound paclitaxel with or without CC-486 as second-line treatment for advanced nonsquamous non-small cell lung cancer (NSCLC). Cancer. 2018;124(24):4667–75.
Tawbi HA, Beumer JH, Tarhini AA, Moschos S, Buch SC, Egorin MJ, et al. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: a phase I/II study and pharmacokinetic analysis. Ann Oncol. 2013;24(4):1112–9.
Sohal D, Krishnamurthi SS, Tohme R, Shepard DR, Khorana AA, Saunthararajah Y. A pilot clinical trial of p53/p16-independent epigenetic therapy for pancreatic ductal adenocarcinoma (PDA). J Clin Oncol. 2020;38(4_suppl):699.
Albany C, Spinella MJ, Adra N, Hanna NH, Einhorn L. A phase I study of guadecitabine (SGI-110) plus cisplatin in patients with platinum refractory germ cell tumors. J Clin Oncol. 2020;38(6_suppl):408.
Lee V, Wang J, Zahurak M, Gootjes E, Verheul HM, Parkinson R, et al. A phase I trial of a guadecitabine (SGI-110) and irinotecan in metastatic colorectal cancer patients previously exposed to irinotecan. Clin Cancer Res. 2018;24(24):6160–7.
Balasubramaniam S, Redon CE, Peer CJ, Bryla C, Lee MJ, Trepel JB, et al. Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anticancer Drugs. 2018;29(5):457–65.
Choy E, Ballman K, Chen J, Dickson MA, Chugh R, George S, et al. SARC018_SPORE02: phase II study of mocetinostat administered with gemcitabine for patients with metastatic leiomyosarcoma with progression or relapse following prior treatment with gemcitabine-containing therapy. Sarcoma. 2018;2018:2068517.
Lee EQ, Reardon DA, Schiff D, Drappatz J, Muzikansky A, Grimm SA, et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 2015;17(6):862–7.
Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol. 2012;107(1):133–8.
Gordon SW, McGuire WP 3rd, Shafer DA, Sterling RK, Lee HM, Matherly SC, et al. Phase I study of sorafenib and vorinostat in advanced hepatocellular carcinoma. Am J Clin Oncol. 2019;42(8):649–54.
Yoo C, Ryu MH, Na YS, Ryoo BY, Lee CW, Kang YK. Vorinostat in combination with capecitabine plus cisplatin as a first-line chemotherapy for patients with metastatic or unresectable gastric cancer: phase II study and biomarker analysis. Br J Cancer. 2016;114(11):1185–90.
Aggarwal R, Abida W, Schweizer M, Pantuck A, Nanus D, Heath E, et al. Abstract CT095: A Phase Ib/IIa study of the BET bromodomain inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res. 2019;79(13 Supplement):CT095.
Lee JJ, Sun W, Bahary N, Ohr J, Rhee JC, Stoller RG, et al. Phase 2 study of pembrolizumab in combination with azacitidine in subjects with metastatic colorectal cancer. J Clin Oncol. 2017;35(15_suppl):3054.
Medicine NLo. Safety and efficacy study of CC-486 with MK-3475 to treat locally advanced or metastatic non-small cell lung cancer. 2015 [updated December 23, 2020. NCT02546986]. https://clinicaltrials.gov/ct2/show/NCT02546986.
Hellmann MD JP, Opyrchal M, Hafez N, Raez LE,, Gabrilovich D WF, Ordentlich P, Brouwer S, Sankoh S, Schmidt E,, Meyers ML RS, editors. Efficacy/safety of entinostat (ENT) and pembrolizumab (PEMBRO) in NSCLC patients previously treated with anti-PD-(L)1 therapy. . IASLC 19th World Conference on Lung Cancer; 2018 September 23–26; Toronto, Canada.
Khushalani NI, Markowitz J, Eroglu Z, Giuroiu I, Ladanova V, Reiersen P, et al. A phase I trial of panobinostat with ipilimumab in advanced melanoma. J Clin Oncol. 2017;35(15_suppl):9547.
Rodriguez CP, Wu QV, Voutsinas J, Fromm JR, Jiang X, Pillarisetty VG, et al. A phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin Cancer Res. 2020;26(4):837–45.
Acknowledgements
We thank Dr. Ryan Johnson for assistance with figure preparation and proofreading of the manuscript.
Funding
This literature review was funded through institutional stipends to the authors.
Author information
Authors and Affiliations
Contributions
NJ was responsible for conceptualization, figure generation, and manuscript drafting. TG was responsible for table generation and manuscript drafting. EB and KH were responsible for conceptualization and coordination. All authors were responsible for reviewing, editing, and approving the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, N., George, T.L., Otterson, G.A. et al. Advances in epigenetic therapeutics with focus on solid tumors. Clin Epigenet 13, 83 (2021). https://doi.org/10.1186/s13148-021-01069-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01069-7