
Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is an age-related disease featured with abnormal fibrotic response and compromised lung function. Cellular senescence is now considered as an essential driving mechanism for IPF. Given the poor knowledge of the mechanisms underpinning IPF progression, understanding the cellular processes and molecular pathways is critical for developing effective therapies of IPF.
Methods
Lung fibrosis was induced using bleomycin in C57BL/6 mice. Cellular senescence was measured by immunofluorescence. The effects of FGF-4 on fibroblast activation markers and signaling molecules were assessed with western blot and qPCR.
Results
We demonstrated elevated abundance of senescent mesenchymal stem cells (MSCs) in IPF lung tissues, which was tightly correlated with the severity of pulmonary fibrosis in vivo. In addition, senescent MSCs could effectively induce the phenotype of pulmonary fibrosis both in vitro and in vivo. To further confirm how senescent MSCs regulate IPF progression, we demonstrate that FGF-4 is significantly elevated in senescent MSCs, which can induce the activation of pulmonary fibroblasts. In vitro, FGF-4 can activate Wnt signaling in a FOXM1-dependent manner. Inhibition of FOXM1 via thiostrepton effectively impairs FGF-4-induced activation of pulmonary fibroblast and dramatically suppresses the development of pulmonary fibrosis.
Conclusion
These findings reveal that FGF-4 plays a crucial role in senescent MSCs-mediated pulmonary fibrogenesis, and suggests that strategies aimed at deletion of senescent MSCs or blocking the FGF-4/FOXM1 axis could be effective in the therapy of IPF.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a is a quintessential disease of aging with median diagnosis at 66 years, characterized by diffuse alveolitis, alveolar structural disorder, and eventually pulmonary interstitial fibrosis [1]. The clinical progress of IPF is usually complicated by acute episodes of respiratory function deterioration and no effective treatments are available in preventing the acute exacerbation of IPF [2]. Given the mechanisms involved in the pathogenesis of IPF remain elusive, investigation on the cellular process and molecular pathways involved are critical for devising effective IPF therapies. Recently, it was reported that cellular senescence has been increasingly recognized as a key contributor to aging and aging-related diseases, including IPF. Thus, better defining the mechanisms of how senescent cells regulating the pathogenesis of IPF may provide a novel strategy for IPF treatment.
Cellular senescence is a state of permanent growth arrest accompany with stereotyped phenotypic changes [3]. Senescent cells could secrete various factors, including proinflammatory cytokines, chemokines and extracellular matrix proteases, which collectively constitute the senescence-associated secretory phenotype (SASP) [4]. Through secreting SASP, senescent cells contribute cell proliferation and tissue deterioration via paracrine mechanism [5]. Therapeutic interventions that ameliorate the burden of senescent cells effectively suppress the progression of pulmonary fibrosis [6]. In our work, we found a large amount of senescent mesenchymal stem cells (MSCs) in fibrotic lung tissues. MSCs are multipotent cells able to self-renew and differentiate into other cell types. MSCs are endogenous MSCs in lungs, which are responsible for maintenance of lung homeostasis. Previous studies showed that MSCs would differentiate into a variety of cell types to promote tissue regeneration [7]. In addition, the paracrine capacity of MSCs could also function in the normal tissue and the pathological state [8, 9]. Recently, increasing evidence has shown that senescent MSCs play a degenerative role in many diseases, especially age-associated diseases [10, 11]. Understanding the negative roles of senescent MSCs in tissue repair and regeneration will benefit the future research to develop therapeutic applications for intervention.
FGF-4 is a member of fibroblast growth factor (FGF) family comprised 18 structurally-related polypeptides. Accumulating evidence suggest that FGF signaling is implicated in the development of IPF [12]. FGF signaling regulates a variety of biological processes via controlling proliferation, differentiation, migration, and metabolism of target cells [13]. In our study, we found FGF-4 was dramatically upregulated in senescent MSCs, which could induce the activation of pulmonary fibroblasts. Aberrantly activated fibroblasts were the main source of increased extracellular matrix in the progression of pulmonary fibrosis [14]. Recent studies have identified potential targets for therapeutic interventions [15], which likely involves the reversion of activated fibroblasts to a quiescent phenotype [16,17,18]. Failure of fibroblasts to reverse to a quiescent phenotype promotes excessive production and remodeling of extracellular matrix (ECM) which characterizes the development of fibrotic disease [19,20,21] and impulses disease progression to end-organ failure and death [22].
To further uncover the underlying mechanism of how FGF-4 induce pulmonary fibroblast activation, we found that FGF-4 could elevate Forkhead Box M1 (FOXM1) expression which could bind with β-catenin and further induce the activation of Wnt signaling. FOXM1, as a member of the Forkhead Box transcription factor family that commonly overexpressed in most human tumors, plays important roles in mammal development, DNA repair, monocyte/macrophage recruitment, and human tumorigenesis [23,24,25,26]. Recently, FOXM1 was reported to be a novel component of Wnt signaling [27], which was elevated in biopsy samples from human patients with interstitial pulmonary fibrosis [28]. In our study, we demonstrated that FOXM1 was significantly elevated in pulmonary fibroblasts of fibrotic lung tissues. Inhibition of FOXM1 through thiostrepton could effectively suppress FGF-4-induced activation of pulmonary fibroblasts, and impaired the development of pulmonary fibrosis. Taken together, our work provides a novel potent mediator of fibrotic event, supporting a potential strategy of regulating FGF-4-mediated fibroblast activation for the treatment of pulmonary fibrosis.
Materials and methods
Ethics statement
The animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals (The Ministry of Science and Technology of China, 2006), and all experimental protocols were approved under the animal protocol number IACUC-2,003,135 by the Animal Care and Use Committee of Nanjing University. The lung tissues of IPF patients (n = 6, male, over 60 years old) were obtained from the Department of Lung Transplantation, Wuxi People’s Hospital, Wuxi, China. Our study was officially approved by the Ethics Committee of Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School.
Cell isolation and cell culture
Male C57BL/6 mice aged 4–6 weeks were sacrificed by cervical dislocation. The lung tissues from the mice were cut into pieces and digested in mixture of 0.2% collagenase I (SCR103, Sigma-Aldrich, St. louis, MO) and 0.2% dispase (D4693, Sigma) for 1 h at 37℃ with shaking. The suspension was filtered through 100- and 40-µm filters and RBC lysis buffer (sc-296258, Santa Cruz, CA, USA) was added to remove red blood cells after centrifuging. Cells were resuspended in MACS buffer (Miltenyi Biotec, Germany) and sorted by AutoMACS cell separator system (Miltenyi Biotec) using CD45 and Sca-1 magnetic beads antibodies. MSCs were cultured in DMEM containing 20% MSC fetal bovine serum (Gibco, Grand Island, NY) in incubator at 37 °C and 5% CO2. Meanwhile, the sedimentary tissue was rinsed with PBS for three times and cultured in DMEM (WISENT, Nanjing, China) containing 10% fetal bovine serum (Gibco). The cells were detached with Trypsin-EDTA (Gibco) and split in new medium when reaching approximately 90% confluence.
To investigated the effects of senescent MSCs on the activation of pulmonary fibroblasts, cell culture inserts (0.4 μm PET, 4.5 cm2, Millipore) were used to establish an indirect co-culture system. MSCs were pre-treated with bleomycin (BLM) accompanied with or without DQ treatment for 24 h in the upper chamber. Then, pulmonary fibroblasts were plated in the lower chamber to co-culture with MSCs for another 2 days. The inserts were removed and pulmonary fibroblasts were harvested for further study.
Senescent cell-induced pulmonary fibrosis
MSCs were treated with 5 µg/ml BLM for 24 h and labelled with DiI by staining for 12 min at 37 °C using Cell Plasma Membrane Staining Kit with DiI (C1991S, Beyotime, Shanghai, China). The labelled MSCs were digested and resuspended at 1 × 106 cells/ml with 50 µl PBS for each mouse. Male C57BL/6 mice were maintained under standard conditions with free access to water and laboratory rodent food for a week and then administered labelled senescent MSCs intratracheally. Control group was injected with normal MSCs. The mice were sacrificed at day 14 after MSCs administration, and lung tissues were collected for further analysis.
Induction and treatment of pulmonary fibrosis
Male C57BL/6 mice (n = 6 each group) were acclimated to the environment for 1 week followed by intratracheally injection of 5 mg/kg BLM dissolved in 50 µl of saline. The control group was injected with 50 ul saline only. 5 mg/kg Dasatinib plus 50 mg/kg Quercetin (DQ, Solarbio, Beijing, China) or vehicle was injected intraperitoneally every two days from day 5 for 2 weeks. Mice were sacrificed at day 19, and lung tissues were collected for further analysis. To explore the role of FOXM1 in pulmonary fibrosis, mice were treated with thiostrepton (HY-B0990, MedChem Express, San Diego, CA), which was known as an inhibitor of FOXM1 [29]. Thiostrepton (30 mg/kg) or vehicle was injected intraperitoneally every other day from 2 weeks. Then, Mice were sacrificed and lung tissues were collected for further analysis.
Histology
Lower right lung lobes were fixed in 4% neutral phosphate-buffered paraformaldehyde overnight, dehydrated, transparentized and embedded in paraffin before sectioning into 5 μm-thick slices. The lung sections were stained with haematoxylin and eosin (H&E) for observation of structure or used for detection of collagen deposition by Masson’s trichrome staining. Tissue area was measured using Image J. Collagen deposition was quantified using the ratio between collagen area and total area. Results were normalized using control group.
Immunofluorescence
The immunofluorescence analysis of lung tissues or cells were performed as described previously. Briefly, samples were permeabilized with 0.3% Triton X-100 and further blocked with goat serum for 1 h. Then, each sample was incubated with primary antibody overnight at 4 °C. The detail information of primary antibodies was listed in Table S2. After washing 3 times, samples were further incubated with fluorophore-labeled secondary antibody (Invitrogen) for 1 h at 37 °C. Nuclei were stained with DAPI (Sigma). The images were captured using a confocal fluorescence microscope (Olympus, Tokyo, Japan). Fluorescence intensity was quantified using Integrated Density (IntDen). Results were normalized using control group. And the colocalization levels was quantified using Manders’ overlap coefficient (MOC).
Immunohistochemistry
Paraffin-embedded 5 μm slides were deparaffinized with xylene before rehydration using an ethanol gradient, followed by antigen retrieval. Next, quenching of endogenous peroxidase activity was achieved by incubation with 3% H2O2 for 10 min. Tissue sections were blocked with goat serum and further incubated with indicated antibodies overnight at 4 °C. After washing 3 times, the lung sections were incubated with the horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h (BOSTER, Wuhan, China). Then, the DAB Substrate System (AiFang biological) was used to reveal the immunohistochemical staining. The images were captured by using Eclipse Ni-U microscope (Nikon). DAB intensity was quantified using Integrated Density (IntDen). Results were normalized using control group.
Western blot
Proteins were extracted from either cells or lung tissues using RIPA buffer added with protease inhibitor cocktail and phosphatase inhibitor cocktail. Western blot analysis was performed as previous described. Proteins were separated by SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes. Then, the membranes were further incubated with indicated primary antibodies overnight at 4 °C. Species-matched horseradish peroxidase-conjugated IgG was used as secondary antibody. The chromogenic signal intensity was detected using an Odyssey Scanning System (LI-COR, Lincoln, NE). Expression levels of the proteins were quantified by densitometry using ImageJ and normalized to the expression of GAPDH.
Quantitative real-time polymerase chain reaction (Q-PCR)
Total RNA was extracted from cells or lung tissues using RNA-easy isolation reagent (Vazyme, Nanjing, China) according to the manufacturer’s instructions and then subjected to reverse transcription polymerase chain reaction (RT-PCR) using the Easyscript first-strand cDNA synthesis super mix kit (Vazyme). Q-PCR was performed using ChamQ SYBR qPCR Master Mix (Vazyme) on a ViiA 7 Q-PCR system (Applied Biosystems, Waltham, MA). Each sample was run in triplicate and the relative quantification of the expression of the target genes was measured using glyseraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA as an internal control. The sequences of primer pairs used in this assay are listed in Table S1.
Co-immunoprecipitation (Co-IP) and protein analysis
Co-IP was performed using the Thermo Scientific Pierce Co-IP kit following the manufacture’s protocol. Briefly, the FOXM1 antibody was first immobilized for 2 h using AminoLink Plus Coupling Resin. The resin was then washed and incubated with the lysate of lung tissues overnight. A negative control that was provided with the IP kit to assess nonspecific binding received the same treatment as the Co-IP samples. Immunocomplexes were washed five times with washing buffer before being resolved by SDS–PAGE and immunoblotted with the indicated antibodies [30].
Statistical analysis
Results are presented as the mean ± SD. Statistical significances were calculated with Student’s t-test for comparisons between 2 groups and ANOVA for multiple group comparisons as showed in figure legends. P values were considered significant at *P < 0.05, **P < 0.01.
Results
Senescent MSCs is tightly correlated with the progression of pulmonary fibrosis
IPF is an age-associated disease, and cellular senescence is reported to be a key mechanism that drive the progression of IPF [31]. Hence, we first determined the senescent circumstance in lungs of IPF patients. As compared with normal lung tissue, p16INK4a and p21Waf1/cip1, as typical markers of cellular senescence, were profoundly increased in fibrotic lung tissues (Fig. 1A, B). Meanwhile, we further demonstrated that the expression of p16INK4a was positive correlated with the levels of COL1A1 in the fibrotic lung tissues of BLM-treated mice (Fig. 1E). To date, growing evidences have demonstrated that mesenchymal stem cells (MSCs) could exert multiple protective effects on pulmonary fibrosis including suppressing collagen deposition and differentiating into local cell types [32]. Interestingly, we found an elevated accumulation of MSCs (Sca-1 positive cells) in fibrotic lung tissues (Fig. S1), which was largely senescent in the progression of pulmonary fibrosis (Fig. 1C, D). In addition, the aggravated pulmonary fibrosis that indicated by Ashcroft score were also tightly correlated with enhanced MSCs senescence in vivo (Fig. 1F). These results indicated that the number of senescent MSCs (Sn-MSCs) was positively correlated with the severity of pulmonary fibrosis.

MSCs senescence is tightly correlated with the progression of pulmonary fibrosis. (A, B) Repentative images and quantification of immunostaining for p16INK4a (A) and p21waf1/cip1 (B) in the lung tissues from patients with IPF. n = 5, **P < 0.01. (C) Representative images and quantification of coimmunostaining of Sca-1 and p16INK4a in lung tissues from patients with IPF. n = 5, **P < 0.01. (D) Representative images of coimmunostaining of Sca-1 and p21waf1/cip1 in lung tissues from patients with IPF. n = 5, **P < 0.01. (E) Correlation curves between relative mRNA expression of p16INK4a and COL1A1 in the lung tissues derived from mice treated with bleomycin. R2 = 0.91396. (F) Correlation curves between senescent MSCs and Ashcroft Score, R2 = 0.94471. Senescent MSCs was represented by Manders’ overlap coefficient (MOC) of p16INK4a and Sca-1
Clearance of senescent MSCs could effectively suppress the development of pulmonary fibrosis
As increased Sn-MSCs were tightly correlated with the severity of pulmonary fibrosis, we further investigated whether elimination of Sn-MSCs could effectively impair the development of pulmonary fibrosis in vivo. Here, dasatinib plus quercetin (DQ) as a widely reported senolytic drug cocktail was intragastrically administrated to BLM-induced pulmonary fibrosis mice. In vitro, treatment with DQ robustly retarded BLM-induced MSCs senescence, as demonstrated by decreased numbers of SA-β-gal positive MSCs (Fig. 2A) and diminished levels of p16INK4a and p21Waf1/Cip1 (Fig. 2B and C). In vivo, body weight monitoring showed that the weight of DQ-treated mice was heavier than single BLM-treated mice when sacrificed (Fig. S2). In addition, DQ treatment could effectively mitigate pulmonary fibrosis as displayed with decreased collagen deposition and a well-preserved lung structure (Fig. 2D). In comparison with BLM-treated mice, immunofluorescence results showed DQ administration obviously decreased the numbers of Sn-MSCs in fibrotic lungs (Fig. 2E and S3A). In addition, the protein levels of fibrotic markers and cellular senescence markers were also dramatically reduced as compared with that in the lung tissues of BLM-treated mice (Fig. 2D, F and S3B). The results indicated that DQ could remove Sn-MSCs, and subsequently attenuate the progression of pulmonary fibrosis.

Clearance of senescent MSCs could effectively suppress the development of pulmonary fibrosis. (A) The representative images of SA-β-gal staining for bleomycin (BLM)-treated MSCs accompany with or without DQ (200nM Dasatinib + 50µM Quercetin) treatment. (B) Western blot analysis of p16INK4a and p21Waf1/cip1 expression in BLM-treated MSCs followed with or without DQ treatment. n = 3, *P < 0.05, **P < 0.01. (C) Immunofluorescence analysis of p16INK4a and p21Waf1/cip1 expression in BLM-treated MSCs followed with or without DQ treatment. n = 5, **P < 0.01. (D-F) Mice (n = 6 in each group) were intraperitoneally injected with vehicle (corn oil) or DQ (5 mg/kg Dasatinib + 50 mg/kg Quercetin) every two days starting 5 days after administration of bleomycin (BLM, 5 mg/kg). (D) Haematoxylin and eosin (H&E) staining, Masson’s trichrome staining and p16INK4a immumohistochemical staining of representative lung sections from mice injected with or without DQ. n = 5, **P < 0.01. (E) The representative images of double fluorescent immunostaining for Sca-1 and p16INK4a in the lung tissues from mice injected with or without DQ. n = 5, **P < 0.01. (F) Western blot analysis of COL1A1, α-SMA, p16INK4a and p21Waf1/cip1in lungs of mice injected with or without DQ. n = 3, *P < 0.05, **P < 0.01
Senescent MSCs could induce pulmonary fibrosis via inducing the activation of pulmonary fibroblasts
In order to confirm the pro-fibrotic role of Sn-MSCs in vivo, mice were intratracheally injected with Sn-MSCs. In vivo, we observed an accumulation of exogenous senescent MSCs (Sn-MSCs) that labeled with DiI in the fibrotic regions (Fig. 3A), along with an increased extent of lung damage and collagen deposition in the lung tissues of mice injected with Sn-MSCs (Fig. 3C). In addition, Sn-MSCs injection dramatically enhanced the levels of fibrotic markers, including α-SMA, COL1A1 (Fig. 3B) and hydroxyproline (Fig. S4), which confirmed the profibrotic role of senescent MSCs in the progression of pulmonary fibrosis. To further investigate how Sn-MSCs promote the development of pulmonary fibrosis, pulmonary fibroblasts were co-cultured with Sn-MSCs pretreated with or without DQ in vitro (Fig. 3D). In the co-culture system, Sn-MSCs could induce the expression of Collagen I and α-SMA (Fig. 3E, F), accompany with enhanced migration ability of pulmonary fibroblasts (Fig. 3G). Oppositely, elimination of senescent MSCs by DQ could profoundly prevent pulmonary fibroblasts activation in the co-culture system (Fig. 3D-G). Our results indicated that eliminating Sn-MSCs could suppress the activation of pulmonary fibroblasts in vitro.

Senescent MSCs could induce pulmonary fibrosis in vivo. (A-C) Mice (n = 6 in each group) were intraperitoneally injected with senescent MSCs (Sn-MSCs injection) or normal MSCs (Con-MSCs injection). (A) Representative DiI fluorescence images for the detection of exogenous MSCs in lung tissues. (B) Western blot analysis for the protein expression of COL1A1 and α-SMA in lungs of mice injected with Sn-MSCs or Con-MSCs. n = 3. *P < 0.05. (C) Haematoxylin and eosin (H&E) staining and Masson’s trichrome staining of representative lung sections from mice injected with Sn-MSCs or Con-MSCs. n = 5, **P < 0.01. (D) Schematic diagram of the pre-treated-MSCs and fibroblasts co-culture system. (E) Western blot analysis of COL1A1 and α-SMA in pulmonary fibroblasts co-cultured with MSCs after indicated treatment. n = 3, *P < 0.05, **P < 0.01. (F) Immunofluorescence analysis of COL1A1 and α-SMA in pulmonary fibroblasts co-cultured with MSCs after indicated treatment. n = 5, **P < 0.01. (G) Migration ability analysis of pulmonary fibroblasts co-cultured with MSCs after indicated treatment. n = 3, **P < 0.01
FGF4 was significantly elevated in Sn-MSCs
It was documented that the secretome called senescence-associated secretory phenotype (SASP) mediated many pathophysiological effects associated with senescent cells. To confirm how Sn-MSCs induce the activation of pulmonary fibroblasts in the development of pulmonary fibrosis, we analyzed the mRNA levels of SASP related genes. Among the 13 genes, FGF-4, IL-4, IL-1β were especially overexpressed in Sn-MSCs (Fig. 4A). In addition, we measured the expression of FGF-4, IL-4 and IL-1β in the supernatant of Sn-MSCs treated with or without DQ in vitro. Our results showed that the concentration of FGF-4 was dramatically increased in the supernatant of Sn-MSCs, and the FGF-4 secretion ability of Sn-MSCs was effectively suppressed with the treatment of DQ (Fig. 4B). In vivo, we also demonstrated that the bronchoalveolar fluid (BALF) derived from BLM-treated mice contained higher concentration of FGF-4 (Fig. S5). The colocalization results showed that FGF-4 was highly expressed in Sca-1 positive cells in both BLM-induced and Sn-MSCs-induced pulmonary fibrosis models (Fig. 4C, E), as compared with in α-SMA or SPC positive cells (Fig. 4D, F, S6). Moreover, we also found that in the lungs of IPF patients, FGF-4 expression was significantly increased and co-localized with fibroblasts (Fig S7). These results indicated that FGF-4 was highly expressed in the MSCs from fibrotic lung tissues.

FGF-4 was significantly elevated in senescent MSCs. (A) Q-PCR analysis of mRNA levels for SASP-related genes (Fgf2, Fgf4, Hbefg, Il1b, Il4, Il6, Tgfb1, Tgfb2, Tgfb3 and Tnfa) in MSCs treated with bleomycin (BLM). n = 3, **P < 0.01. (B) The secretion of IL-4, IL-1β and FGF-4 in the supernatant of BLM-treated MSCs followed with or without DQ (200nM Dasatinib + 50µM Quercetin) treatment. n = 3, **P < 0.01. (C) Repentative images of double immunofluorescent staining of Sca-1 and FGF-4 in the lung tissues of mice treated with or without BLM. (D) The quantification of immunofluorescence colocalization levels of MSCs, fibroblasts and epithelial cells with FGF-4 in the lung tissues of mice treated with or without BLM via using Manders’ overlap coefficient (MOC). n = 3, **P < 0.01. (E) Repentative images of double immunofluorescent staining of Sca-1 and FGF-4 in the lung tissues of mice injected with Con-MSCs or senescent MSCs (Sn-MSCs). (F) The quantification of immunofluorescence colocalization levels of MSCs, fibroblasts and epithelial cells with FGF-4 in the lung tissues of mice injected with Con-MSCs or Sn-MSCs. n = 3, **P < 0.01
Inhibiting FGF-4 could attenuate Sn-MSCs-induced activation of pulmonary fibroblasts
The FGF family was mainly characterized by the ability of its members to promote fibroblast proliferation [33], which has been reported to be implicated in the pathogenesis of IPF [34]. To further confirm the role of FGF-4 in Sn-MSCs-induced pulmonary fibroblast activation, MSCs were transfected with the siRNA of FGF-4 (si-FGF-4) accompany with or without the treatment of BLM (Fig. 5A). Interestingly, knockdown of FGF-4 significantly impaired Sn-MSCs-induced activation of pulmonary fibroblasts, as evidenced by decreased expression of myofibroblast markers (Collagen I and α-SMA) in the co-culture system (Fig. 5B-D). In addition, we further proved that dampening the expression of FGF-4 in Sn-MSCs could effectively suppress the migration ability of pulmonary fibroblasts that promoted by Sn-MSCs (Fig. 5E). These results indicated that FGF-4 was required for Sn-MSCs-induced activation of pulmonary fibroblast in vitro.

Inhibiting FGF-4 could attenuate Sn-MSCs-induced activation of pulmonary fibroblasts. (A) Q-PCR analysis of mRNA levels of FGF-4 in MSCs transfected with or without si-FGF-4. n = 3, *P < 0.05. (B) Schematic diagram of cell co-culture system with pre-treated MSCs and fibroblasts. (C) Western blot analysis of COL1A1 and α-SMA expression in pulmonary fibroblasts co-cultured with MSCs transfected with or without si-FGF-4. n = 3, *P < 0.05, **P < 0.01. (D) Immunofluorescence analysis the expression of COL1A1 and α-SMA in pulmonary fibroblasts co-cultured with MSCs transfected with or without si-FGF-4. n = 5, **P < 0.01. (E) Migration ability analysis of pulmonary fibroblasts co-cultured with MSCs after indicated treatment. n = 3, *P < 0.05, **P < 0.01
FGF-4 could induce the activation of pulmonary fibroblasts via FOXM1/β -catenin axis
Given that Wnt signaling played a pivotal role in fibroblast activation, we next investigated the effect of FGF-4 on pulmonary fibroblasts activation. FGF-4 could increase the mRNA levels of α-SMA, COL1A1 (Fig. 6A) and ECM related genes (Fig. 6B), along with elevated levels of Wnt target genes, including cyclin D1, β-catenin and Axin2 (Fig. 6A, C, D). FGF-4 also promoted the nuclear translocation of β-catenin in pulmonary fibroblasts as determined by immunofluorescence analysis (Fig. 6E). These results indicated that FGF-4 may promote the myofibroblast differentiation of fibroblasts via activating Wnt signaling. To further confirm the role of Wnt signaling in FGF-4-induced activation of pulmonary fibroblasts, Wnt signaling was silenced by ICG-001 which could bind cAMP-responsive element binding (CREB)-binding protein (CBP) to disrupt its interaction with β-catenin [35]. Our results demonstrated that impairing Wnt signaling could remarkably suppress FGF-4-induced myofibroblast differentiation and migration ability of pulmonary fibroblasts (Fig. 6C-F), which suggested that FGF-4 could activate pulmonary fibroblasts through activating Wnt signaling.

FGF-4 could induce the activation of pulmonary fibroblasts via activating Wnt signaling. (A) Q-PCR analysis of mRNA levels of Cyclin D1, Axin2, β-catenin, Col1a1 and α-SMA in pulmonary fibroblasts treated with or without FGF-4 (1 ng/ml). n = 3, **P < 0.01. (B) Q-PCR analysis of mRNA levels of Acan, Eln, FN1, and Lama1 in pulmonary fibroblasts treated with or without FGF-4 (1 ng/ml). n = 3, *P < 0.05, **P < 0.01. (C) Western blot analysis of COL1A1, β-catenin, and α-SMA expression in FGF-4-treated pulmonary fibroblasts followed with the treatment of ICG-001 (10 nM) or not. n = 3, *P < 0.05, **P < 0.01. (D) Immunofluorescence staining and quantification of COL1A1 and α-SMA in pulmonary fibroblasts treated as in B. n = 5, **P < 0.01. (E) Immunofluorescence analysis the expression of β-catenin in pulmonary fibroblasts treated as in B. n = 5, **P < 0.01. (F) The migration ability assay of pulmonary fibroblasts treated as in B. n = 3, **P < 0.01
To elucidate the molecular pathway involved in FGF-4-induced Wnt signaling activation, we focused on FOXM1 that was reported to be critical for β-catenin nuclear localization and transcriptional function [36]. Here, we performed co-staining on lung sections of donors and IPF patients, and found that FOXM1 expression was elevated in fibroblasts as demonstrated by colocalization with α-SMA, a marker of fibroblasts (Fig. 7A, Fig. S8A), which was further confirmed in BLM-induced mouse pulmonary fibrosis models (Fig. 7B, C, Fig. S8B, C). In vitro, FGF-4 could profoundly increase the expression of FOXM1 (Fig. 7D), resulting the nuclear translocation of FOXM1 (Fig. 7F, Fig. S8D, E) and enhanced binding ability of FOXM1 with β-catenin (Fig. 7E). Oppositely, inhibition of FOXM1 by thiostrepton could effectively suppress the activation of pulmonary fibroblasts, as confirmed by decreased expression of α-SMA and COL1A1 (Fig. 7D, G, Fig. S8F), and impaired migration ability (Fig. 7H). In vivo, we investigated whether silencing FOXM1 could block the development of pulmonary fibrosis. Compared with BLM-treated mice, thiostrepton evidently decreased the extent of lung lesions and attenuated collagen deposition (Fig. 7I, Fig. S8G, H). Immunolabeling also showed that FOXM1 inhibition profoundly attenuated the expression of fibrotic markers α-SMA and collagen I (Fig. 7J, K, Fig. S8I, J), along with the inhibition of Wnt/β-catenin signaling as shown by reduced expression of β-catenin (Fig. 7J, Fig. S8I). In addition, thiostrepton treatment effectively decreased FOXM1 expression in pulmonary fibroblasts of BLM-induced fibrotic mice (Fig. 7L, Fig. S8K). These results demonstrated that inhibition of FOXM1 with thiostrepton could protect mice from BLM-induced pulmonary fibrosis.

FOXM1 plays an essential role in FGF-4-induced pulmonary fibroblasts activation and pulmonary fibrogenesis. (A) Representative images of coimmunostaining of FOXM1 and α-SMA in lung tissues from patients with IPF. (B) Representative images of coimmunostaining of FOXM1 and α-SMA in lung tissues from bleomycin (BLM)-treated mice. (C) Western blot analysis of FOXM1 expression in pulmonary fibroblasts isolated from BLM-treated mice. (D) Western blot analysis of protein levels of COL1A1, FOXM1 and α-SMA in FGF-4-treated pulmonary fibroblasts followed with the treatment of Thiostrepton (Thi, 10 µM). (E) Co-IP analysis of β-catenin-FOXM1 interaction in pulmonary fibroblasts treated as in D. (F, G) Immunofluorescence analysis the expression of FOXM1 (F), α-SMA and COL1A1 (G) in pulmonary fibroblasts treated as in D. (H) The migration ability assay of pulmonary fibroblasts treated as in D. n = 3, **P < 0.01. (I) Haematoxylin and eosin (H&E) staining, Masson’s trichrome staining of representative lung sections (n = 6) from BLM-treated mice injected with or without Thi (30 mg/kg). (J) Western blot analysis of α-SMA, β-catenin and FOXM1 expression in the lung tissues from mice treated as in I. (K) Representative images of coimmunostaining of α-SMA and COL1A1in lung tissues from mice treated as in I. (L) Representative images of coimmunostaining of FOXM1 and α-SMA in lung tissues from mice treated as in I
Discussion
Pulmonary fibrosis is a refractory and fatal disease that carries a high mortality rate currently. Existing treatments for pulmonary fibrosis could not improve the survival significantly, resulting an urgent requirement for new approaches in clinical therapy [37]. Recently, cellular senescence has been demonstrated to be essential in contributing fibrotic lung diseases and can be targeted to improve pulmonary function [6]. Here, we show that senescence effectors and molecular markers significantly increase in MSCs of individuals with IPF, which could be efficiently killed by a senolytic cocktail treatment. To define the underlying mechanisms of how senescent MSCs contribute to the initiation and progression of IPF, we screened the expression of SASP components, and found FGF-4 was dramatically increased in senescent MSCs. In addition, we confirmed that senescent MSCs-secreted FGF-4 could induce fibroblast activation, partially clarifying the interaction between senescent MSCs and fibroblasts.
Pulmonary fibrosis was characterized by the persistence of activated fibroblasts which contribute to the deposition of ECM and profound tissue remodeling [38]. In the process of fibrotic lung remodeling, different cellular origins of fibroblasts contribute to fibrotic lesions, including proliferation of resident lung fibroblasts, differentiation of epithelial cells into fibroblasts and recruitment of circulating progenitor fibrocytes from bone marrow [39,40,41]. To date, great efforts have been made to uncover the underlying mechanisms utilized by fibroblasts for survival, proliferation and persistent activation. Recently, there were evidence that epithelial cells differentiated into FOXM1 positive fibroblasts in the progression of pulmonary fibrosis [28]. In addition, FOXM1 could promote fibroblasts differentiate into profibrotic α-SMA+ myofibroblasts [42, 43]. Fully activated fibroblasts were highly contractile cells that remodel surrounding ECM [44]. So, whether FOXM1 played an essential role in FGF-4-induced activation of fibroblasts and the progression of pulmonary fibrosis? In the present study, we investigated the role of FOXM1 in pulmonary fibrosis and the effects of its modulation on the development of pulmonary fibrosis. We detected an extensive increase of FOXM1 expression in human pulmonary fibrosis samples compared with donors. Similarly, the level of FOXM1 was increased during BLM-induced pulmonary fibrosis. Stimulation of mouse pulmonary fibroblasts with FGF-4 dramatically enhanced FOXM1 expression, with the activation of Wnt/β-catenin signaling, elevated proliferation and strengthened migration. Whereas, targeted inhibiting the activity of FOXM1 evoked antifibrotic responses in vitro on mouse pulmonary fibroblasts. These findings provided the foundation for the exploitation of new therapeutic approaches based on the inhibition of FOXM1.
FOXM1 belongs to the forkhead box transcription factor family, which plays an important role in regulating embryonic development, carcinogenesis and organ regeneration via promoting cell cycle progression [45]. During cell cycle, FoxM1 is expressed at late G1 and early S phase, sustained throughout G2 phase and mitosis, and its activity is regulated via phosphorylation [46,47,48]. Expression of FoxM1 protein is low in quiescent cells. Aberrantly overexpressed of FOXM1 have been implicated in several lung diseases [49, 50]. Conditional knockouting the FOXM1 gene or suppressing the activity of FOXM1 protein in bronchiolar progenitor cells could protect mice from allergic responses, resulting reduced mucus hyperplasia and lung inflammation [51]. Thiostrepton was ever reported to be a proteasomal inhibitor that prevents FOXM1 from binding and activating its own promoter [52]. We demonstrated that diminished nuclear localization of FOXM1 could be a reason of decreased Wnt/β-catenin signaling, as the expression of β-catenin that bind with FOXM1 was extensively impaired. In vivo, thiostrepton effectively counteracted BLM-induced pulmonary fibrosis through inhibition of FOXM1 nuclear localization. Although, pirfenidone and nintedanib are currently clinically approved and available for IPF treatment, there are some limitations in side effects and effectiveness [53]. Therefore, exploring new strategies to address the unmet therapeutic need of lung fibrosis is necessary, and the development and clinical application of novel FOXM1-targeted inhibitors will provide a promising future for overcoming IPF.
Conclusions
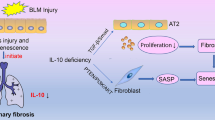
In conclusion, our work implicated the critical role of senescent MSCs in pulmonary fibrogenesis, and revealed that senescent MSCs regulate pulmonary fibroblast activation in FGF-4-dependent manner. In addition, we demonstrated that FGF-4 could promote the expression of FOXM1 which further induce the activation of Wnt signaling via binding with β-catenin. Suppression of FOXM1 by thiostrepton could effectively impair FGF-4-induced activation of pulmonary fibroblasts (Fig. 8).

Schematic model for the crosstalk between senescent MSCs and pulmonary fibroblasts
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–61.
Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr., Lasky JA, Loyd JE, Noth I, Olman MA, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–43.
Tominaga K. The emerging role of senescent cells in tissue homeostasis and pathophysiology. Pathobiol Aging Age Relat Dis. 2015;5:27743.
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46.
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532.
Samsonraj RM, Raghunath M, Nurcombe V, Hui JH, van Wijnen AJ, Cool SM. Concise Review: multifaceted characterization of human mesenchymal stem cells for Use in Regenerative Medicine. Stem Cells Transl Med. 2017;6:2173–85.
Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. 2019;117:15–22.
Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114.
Lee BC, Yu KR. Impact of mesenchymal stem cell senescence on inflammaging. BMB Rep. 2020;53:65–73.
Fraile M, Eiro N, Costa LA, Martín A, Vizoso FJ. Aging and mesenchymal stem cells: Basic concepts, challenges and strategies. Biology (Basel) 2022, 11.
Chaudhary NI, Roth GJ, Hilberg F, Müller-Quernheim J, Prasse A, Zissel G, Schnapp A, Park JE. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–85.
Huang P, Stern MJ. FGF signaling in flies and worms: more and more relevant to vertebrate biology. Cytokine Growth Factor Rev. 2005;16:151–8.
Trotsyuk AA, Chen K, Hyung S, Ma KC, Henn D, Mermin-Bunnell AM, Mittal S, Padmanabhan J, Larson MR, Steele SR, et al. Inhibiting fibroblast mechanotransduction modulates severity of idiopathic pulmonary fibrosis. Adv Wound Care (New Rochelle). 2022;11:511–23.
Taggart C, Mall MA, Lalmanach G, Cataldo D, Ludwig A, Janciauskiene S, Heath N, Meiners S, Overall CM, Schultz C et al. Protean proteases: at the cutting edge of lung diseases. Eur Respir J 2017, 49.
Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–21.
Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–53.
Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, Pradere JP, Friedman RA, Schwabe RF. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073–e10831022.
Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis–a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10:390–402.
Rockey DC, Bell PD, Hill JA. Fibrosis–A common pathway to Organ Injury and failure. N Engl J Med. 2015;373:96.
Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–67.
Urban ML, Manenti L, Vaglio A. Fibrosis–A common pathway to Organ Injury and failure. N Engl J Med. 2015;373:95–6.
Kalin TV, Ustiyan V, Kalinichenko VV. Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle. 2011;10:396–405.
Kalin TV, Wang IC, Meliton L, Zhang Y, Wert SE, Ren X, Snyder J, Bell SM, Graf L Jr., Whitsett JA, Kalinichenko VV. Forkhead Box m1 transcription factor is required for perinatal lung function. Proc Natl Acad Sci U S A. 2008;105:19330–5.
Kalinichenko VV, Lim L, Shin B, Costa RH. Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am J Physiol Lung Cell Mol Physiol. 2001;280:L695–704.
Raychaudhuri P, Park HJ. FoxM1: a master regulator of tumor metastasis. Cancer Res. 2011;71:4329–33.
Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. FoxM1 promotes beta-catenin nuclear localization and controls wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–42.
Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013;32:231–44.
Ju SY, Huang CY, Huang WC, Su Y. Identification of thiostrepton as a novel therapeutic agent that targets human colon cancer stem cells. Cell Death Dis. 2015;6:e1801.
Adebiyi A, Narayanan D, Jaggar JH. Caveolin-1 assembles type 1 inositol 1,4,5-trisphosphate receptors and canonical transient receptor potential 3 channels into a functional signaling complex in arterial smooth muscle cells. J Biol Chem. 2011;286:4341–8.
Cho SJ, Stout-Delgado HW. Aging and lung disease. Annu Rev Physiol. 2020;82:433–59.
Zanoni M, Cortesi M, Zamagni A, Tesei A. The role of mesenchymal stem cells in Radiation-Induced Lung Fibrosis. Int J Mol Sci 2019, 20.
Kim KK, Sisson TH, Horowitz JC. Fibroblast growth factors and pulmonary fibrosis: it’s more complex than it sounds. J Pathol. 2017;241:6–9.
Yang L, Zhou F, Zheng D, Wang D, Li X, Zhao C, Huang X. FGF/FGFR signaling: from lung development to respiratory diseases. Cytokine Growth Factor Rev. 2021;62:94–104.
Danieau G, Morice S, Renault S, Brion R, Biteau K, Amiaud J, Cadé M, Heymann D, Lézot F, Verrecchia F et al. ICG-001, an inhibitor of the β-Catenin and cAMP response element-binding protein dependent gene transcription, decreases proliferation but enhances Migration of Osteosarcoma cells. Pharmaceuticals (Basel) 2021, 14.
Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. FoxM1 promotes β-catenin nuclear localization and controls wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–42.
Kolilekas L, Papiris S, Bouros D. Existing and emerging treatments for idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2019;13:229–39.
Buchner M, Park E, Geng H, Klemm L, Flach J, Passegue E, Schjerven H, Melnick A, Paietta E, Kopanja D, et al. Identification of FOXM1 as a therapeutic target in B-cell lineage acute lymphoblastic leukaemia. Nat Commun. 2015;6:6471.
Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–50.
Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8:145–50.
Andersson-Sjoland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, Pardo A, Westergren-Thorsson G, Selman M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–40.
Pratsinis H, Giannouli CC, Zervolea I, Psarras S, Stathakos D, Kletsas D. Differential proliferative response of fetal and adult human skin fibroblasts to transforming growth factor-beta. Wound Repair Regen. 2004;12:374–83.
Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–37.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63.
Chen X, Muller GA, Quaas M, Fischer M, Han N, Stutchbury B, Sharrocks AD, Engeland K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol Cell Biol. 2013;33:227–36.
Chen YJ, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, Lau LF, Raychaudhuri P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem. 2009;284:30695–707.
Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol. 2008;28:3076–87.
Park HJ, Wang Z, Costa RH, Tyner A, Lau LF, Raychaudhuri P. An N-terminal inhibitory domain modulates activity of FoxM1 during cell cycle. Oncogene. 2008;27:1696–704.
Balli D, Zhang Y, Snyder J, Kalinichenko VV, Kalin TV. Endothelial cell-specific deletion of transcription factor FoxM1 increases urethane-induced lung carcinogenesis. Cancer Res. 2011;71:40–50.
Balli D, Ren X, Chou FS, Cross E, Zhang Y, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene. 2012;31:3875–88.
Ren X, Shah TA, Ustiyan V, Zhang Y, Shinn J, Chen G, Whitsett JA, Kalin TV, Kalinichenko VV. FOXM1 promotes allergen-induced goblet cell metaplasia and pulmonary inflammation. Mol Cell Biol. 2013;33:371–86.
Hegde NS, Sanders DA, Rodriguez R, Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem. 2011;3:725–31.
Roach KM, Castells E, Dixon K, Mason S, Elliott G, Marshall H, Poblocka MA, Macip S, Richardson M, Khalfaoui L, Bradding P. Evaluation of Pirfenidone and Nintedanib in a human lung model of Fibrogenesis. Front Pharmacol. 2021;12:679388.
Acknowledgements
Not applicable.
Funding
This work was supported by the Jiangsu Province Key Research and Development Program (BE2021707); National Natural Science Foundation of China (82372459, 82000069, 82300091); Natural Science Foundation of Jiangsu Province (BK20200314); The China Postdoctoral Science Foundation (2024T170409); Jiangsu Funding Program for Excellent Postdoctoral Talent (2022ZB688); State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM2208).
Author information
Authors and Affiliations
Contributions
The authors contributed in the following ways: XC, BS and XDH were involved in the conception and design of the experiments; YXL and XC wrote the manuscript; YXL and XC performed most of the experiments, JJ, and SDZ contributed to some experiments. AW, and DML revised the manuscript. All authors have given final approval of the presented version to be published.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
The study was support by the project of “Study on the molecular mechanism of senescent MSCs-mediated fibroblast activation in IPF”, which was approved by the Ethics Committee of Nanjing university (IACUC-2003135, Date: 2020-3-24). All patients provided written informed consent, and approval was provided by the ethics committee of Drum Tower Hospital of Nanjing (2020-176-13, Date: 2020-3-10).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest in this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Ji, J., Zheng, S. et al. Senescent lung-resident mesenchymal stem cells drive pulmonary fibrogenesis through FGF-4/FOXM1 axis. Stem Cell Res Ther 15, 309 (2024). https://doi.org/10.1186/s13287-024-03866-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-024-03866-2