
Abstract
O-linked-N-acetylglucosaminylation (O-GlcNAcylation) is a common and important post-translational modification (PTM) linking O-linked β-N-acetylglucosamine (O-GlcNAc) to serine and threonine residues in proteins. Extensive research indicates its impact on target protein stability, activity, and interactions. O-linked N-acetylglucosamine transferase (OGT) is a critical enzyme that catalyzes O-GlcNAc modification, responsible for adding O-GlcNAc to proteins. OGT and O-GlcNAcylation are overexpressed in many tumors and closely associated with tumor growth, invasion, metabolism, drug resistance, and immune evasion. This review delineates the biochemical functions of OGT and summarizes its effects and mechanisms in tumors. Targeting OGT presents a promising novel approach for treating human malignancies.
Similar content being viewed by others


Introduction
O-linked-N-acetylglucosaminylation (O-GlcNAcylation), an important protein glycosylation modification, has its origins in the earliest report by Hart’s team [1, 2]. Subsequently, researchers defined O-GlcNAcylation as the process of adding O-linked β-N-acetylglucosamine (O-GlcNAc) to serine or threonine residues in proteins [3]. This modification is implicated in various cellular processes, such as signal transduction, cell cycle regulation, transcriptional control, and metabolism [4,5,6,7].
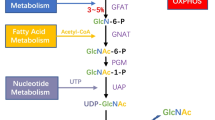
Roughly 2–5% of glucose feeds into the hexosamine biosynthetic pathway (HBP) to generate UDP-GlcNAc, the sugar donor for O-GlcNAcylation [8]. The key rate-limiting enzyme in the HBP pathway is glutamine–fructose-6-phosphate amido transferase (GFAT), which converts fructose-6-phosphate into glucosamine-6-phosphate [9]. O-GlcNAcylation undergoes dynamic and reversible regulation through O-GlcNAc addition and removal. O-GlcNAc transferase (OGT) attaches O-GlcNAc to Ser/Thr residues of substrate proteins, while O-GlcNAcase (OGA) is responsible for its cleavage [10].
The gene responsible for encoding OGT resides on the X chromosome [11]. The OGT protein consists of an N-terminal tetratricopeptide-repeats (TPRs) domain, which binds substrate proteins, and a C-terminal catalytic domain that catalyzes substrate O-GlcNAcylation [12, 13]. Moreover, OGT participates in diverse physiological processes, including fostering nervous system development, regulating mammalian cell physiology, and preserving hematopoietic stem cells. Its expression is elevated in various tumors, suggesting a role in tumor promotion. This review outlines the biochemical functions of OGT and summarizes its role and specific mechanisms in tumors, aiming to provide new insights and approaches for treating malignant tumors.
Structure and basic function of OGT
OGT, a member of the GT-B glycosyltransferase family, is responsible for attaching O-GlcNAc to substrate proteins [14, 15]. OGT is highly conserved across various organisms, from Caenorhabditis elegans to mammals [16]. In humans, alternative splicing produces three OGT isoforms: nucleocytoplasmic OGT (ncOGT), mitochondrial OGT (mOGT), and short OGT (sOGT), each differing in location and length [17]. These isoforms are encoded by the same gene on the X chromosome and feature N-terminal TPRs and a multi-domain catalytic C-terminal [18]. The primary structural difference between the three OGT isoforms is the number of N-terminal TPRs. The longest isoform, ncOGT, contains 13.5 TPRs, while mOGT and sOGT contain 9.5 and 2.5 TPRs, respectively [19, 20]. TPR sequences are primarily involved in the recognition and binding of substrate proteins by OGT [21, 22]. These sequences fold into an antiparallel α-helical structure, with adjacent repeats forming a superhelical structure that binds specific substrates [23, 24]. The enzymatic domain of OGT is located at the C-terminus and catalyzes the O-GlcNAcylation of substrate proteins [12]. This modification affects protein stability, conformation, localization, and activity [25,26,27,28,29]. To date, thousands of proteins have been identified as O-GlcNAcylation targets, including transcription factors, membrane proteins, and cytoskeletal proteins. These modifications regulate gene transcription, cellular responses, protein translation, protein degradation, and other critical biological processes. O-GlcNAcylation impacts cell signal transduction and plays a crucial regulatory role in normal growth and development, as well as in the pathogenesis of various diseases [30,31,32,33].
OGT in cancer progression
Roles of OGT in cancer proliferation
Abnormal proliferation is a hallmark of cancer. The carcinogenic effect of OGT is closely associated with its role in driving cell growth in various malignancies, such as liver cancer, gastric cancer (GC), and colorectal cancer (CRC). [34,35,36]. OGT promotes tumor proliferation primarily through its involvement in regulating protein post-translational modifications (PTMs).
In non-small cell lung cancer cells, OGT overexpression following glutamine deprivation abolishes fructose-1,6-bisphosphatase 1 (FBP1) phosphorylation and enhances β-oxidation gene transcription via FBP1 O-GlcNAcylation, thus promoting cell proliferation [37]. Similar findings have been observed in hepatocellular carcinoma (HCC), highlighting OGT’s crucial role in tumor growth by regulating FBP1 [38]. Additionally, Y box binding protein 1 (YB-1), a well-known oncoprotein, is associated with tumor immune evasion and drug resistance [39, 40]. Liu et al. demonstrated that OGT increases O-GlcNAcylation of YB-1 at Thr126, thereby promoting cell proliferation in HCC [41]. Targeting OGT significantly impedes the progression of high-fructose-induced HCC, with the O-GlcNAcylation of eukaryotic elongation factor 1A1 (EEF1A1) playing a pivotal role in this process [42]. Mitogen-activated protein kinase kinase 2 (MEK2), an important molecule in the MAPK signaling pathway, is related to cell proliferation, differentiation, and stress response [43]. OGT promotes the stability of MEK2 through O-GlcNAcylation at Thr13, thereby enhancing the proliferation and migration of breast cancer cells [44]. Furthermore, microRNA-485-5p modulates CRC proliferation by regulating the stability of B-cell-specific Moloney murine leukemia virus integration region 1 (Bmi-1) via OGT [45]. In xenograft models, mutating the O-GlcNAcylation site of YTH domain family 1 (YTHDF1) reduced tumor growth [46]. DNA polymerase iota (Pol ι) activates glucose-6-phosphate dehydrogenase (G6PD) through Erk-OGT-induced O-GlcNAcylation, promoting the proliferation of esophageal squamous cell carcinoma [47]. Yu et al. found that miR-483 targets OGT to inhibit the proliferation of GC cells [48]. Moreover, the X-inactive-specific transcript (XIST)/miR-424-5p/OGT axis regulates RAF1 glycosylation, impacting liver cancer growth [49]. Long non-coding RNA RHPN1-AS1 is significantly upregulated in CRC cell lines, facilitating CRC progression by modulating the miR-7-5p/OGT axis [50]. These findings indicate that OGT is a key regulator of tumor proliferation (Fig. 1).

OGT and proliferation regulation in cancer
Roles of OGT in cancer invasion and metastasis
Metastasis, the spread and growth of tumor cells from their original site to new locations in the body, is the leading cause of cancer-related deaths [51]. Despite extensive research on invasion and metastasis mechanisms, this regulatory process remains poorly understood. Recent studies have identified OGT as a key regulator of tumor invasion and metastasis.
Epithelial-mesenchymal transition (EMT) is the phenotypic change of cells from an epithelial to a mesenchymal state, resulting in increased motility and invasiveness of tumor cells [52]. EMT is characterized by the loss of epithelial cell-cell connections, cytoskeletal reorganization, decreased expression of E-cadherin, and increased N-cadherin [53]. Numerous studies have explored EMT’s role in promoting tumor cell invasion and malignancy. Jiang et al. found that knocking down enhancer of zeste homolog 2 (EZH2) in colorectal cancer partially reverses the EMT changes induced by OGT-mediated O-GlcNAcylation [54]. Additionally, OGT knockdown has been shown to inhibit the expression of EMT markers (N-cadherin and Slug), migration, and invasion in lung cancer cells, with the interaction between OGT and STAT3 playing a crucial role in this process [55]. Moreover, OGT knockdown in HO-8910PM cells resulted in decreased O-GlcNAcylation and increased expression of E-cadherin [56].
Matrix metalloproteinases (MMPs), members of the metzincin protease superfamily, degrade the extracellular matrix (ECM) [57]. MMPs participate in physiological processes such as embryonic development and wound healing and play a vital role in enhancing tumor cell migration and invasion [58, 59]. OGT regulates matrix metalloproteinase levels, thereby affecting tumor metastasis. Qiao et al. found that suppressing OGT weakened the migration ability of esophageal cancer cells by significantly reducing the expression of matrix metalloproteinase 9 (MMP9) in Eca-109 cells [60]. Furthermore, OGT knockdown in prostate cancer cell lines was associated with reduced expression of MMP-2, MMP-9, and vascular endothelial growth factor (VEGF), thereby inhibiting invasion and angiogenesis through the regulation of the oncogenic transcription factor forkhead box M1 (FoxM1) [61].
OGT orchestrates O-GlcNAcylation to drive the migration and invasion of papillary thyroid cancer by activating Yes-associated protein (YAP) at the Ser109 modification site [62]. Lv et al. identified upregulated OGT expression in HCC, demonstrating its role in promoting tumor aggressiveness through OGT-mediated O-GlcNAcylation, which stabilizes ras-related protein Rab-10 (RAB10) [63]. CD36, a cell membrane protein, mediates fatty acid uptake and is associated with fatty acid absorption in the heart, skeletal muscle, and adipose tissue [64, 65]. Jiang et al. confirmed that fatty acids promote gastric cancer metastasis by inducing CD36 expression via OGT-mediated O-GlcNAcylation [66]. YTH N6-methyladenosine RNA binding protein 2 (YTHDF2) plays a crucial role in N6-methyladenosine (m6A) modification, regulating mRNA degradation [67, 68]. OGT promotes hepatitis B virus-related HCC migration and invasion by mediating O-GlcNAcylation of YTHDF2 at Ser263 [69]. Wang et al. found that reticulon 2 (RTN2) interacts with OGT and is modified by O-GlcNAc; inhibiting OGT abolishes the stimulatory effects of RTN2 on cell migration [70]. SRC-associated in mitosis of 68 kDa (SAM68) is O-GlcNAcylated and predominantly interacts with OGT in the nucleus, promoting lung cancer cell migration and invasion [71]. Multiple studies have linked MORC family CW-type zinc finger 2 (MORC2) with DNA damage and resistance to radiotherapy and chemotherapy in breast cancer [72, 73]. Liu et al. found that OGT O-GlcNAcylates MORC2 at Thr556, thereby promoting breast cancer migration, invasion, and metastasis [74]. Reginato’s team demonstrated that reducing OGT expression significantly decreases FoxM1 protein levels, inhibiting breast cancer cell growth and invasion [75]. Non-coding RNAs also regulate breast cancer progression by altering OGT expression. Inhibition of OGT by miR-24 reduces the stability of forkhead box protein A1 (FOXA1), thereby inhibiting breast cancer cell invasion [76]. Overexpression of OGT significantly enhances O-GlcNAcylation in TAK1 binding protein 3 (TAB3), promoting migration and invasion of triple-negative breast cancer (TNBC) cells in vivo and in vitro [77]. Dysregulation of the NF-κB pathway is increasingly recognized as a key regulator of tumor progression and drug resistance [78, 79]. Ali et al. confirmed that OGT knockdown reduced CXCR4 expression by decreasing O-GlcNAcylation of NF-κB p65 (p65), inhibiting cervical cancer metastasis [80]. Niu et al. showed that OGT-mediated O-GlcNAcylation regulates ras homolog family member A (RhoA) activity in ovarian cancer cells, affecting their migration and invasion [81]. These studies indicate that OGT overexpression significantly promotes tumor cell invasion into other tissues, facilitating their survival and cancer spread. Therefore, targeting OGT may help inhibit tumor metastasis (Fig. 2) [82,83,84,85,86].

OGT in cancer invasion and metastasis regulation
Roles of OGT in cancer metabolism
Abnormal cancer metabolism plays a crucial role in tumorigenesis, metastasis, and drug resistance [87]. Glucose, lipid, and amino acid metabolism in tumor tissue undergo significant changes compared to normal tissue [88]. Studies have shown that OGT is directly or indirectly involved in the regulation of tumor metabolic processes.
Most tumor cells produce adenosine triphosphate (ATP) primarily through glycolysis, even under adequate oxygen levels. This phenomenon, known as the Warburg effect, supports tumor cell growth [89]. Phosphoglycerate kinase 1 (PGK1) is the first ATP-generating enzyme in glycolysis, and its expression is linked to tumor progression [90,91,92]. Research has shown that OGT overexpression enhances PGK1 activity by increasing its O-GlcNAcylation. Blocking T255 O-GlcNAcylation of PGK1 inhibits glycolysis, enhances the mitochondrial tricarboxylic acid (TCA) cycle, and suppresses colon cancer growth [93]. Additionally, transient expression of wild-type OGT increases PKM2 O-GlcNAcylation, suppresses pyruvate kinase activity in HeLa cells, stimulates aerobic glycolysis, and promotes tumor growth [94]. OGT-mediated O-GlcNAcylation also enhances the stability of isocitrate dehydrogenase 2 (IDH2) protein, thereby activating the NF-κB signaling pathway, reprogramming glucose metabolism, and promoting CRC progression [95].
Reprogramming lipid metabolism is a hallmark of many malignancies. Increased fat uptake and lipogenesis occur in various cancers, leading to rapid tumor growth. Lipids form the basic structure of membranes and also serve as signaling molecules and energy sources [96, 97]. Sterol regulatory element-binding protein 1 (SREBP-1) is a major transcription factor controlling lipid metabolism and a key link between oncogenic signaling and tumor metabolism [98]. OGT regulates the expression of SREBP-1 in a proteasomal and AMP-activated protein kinase (AMPK)-dependent manner, thereby altering lipid metabolism and impacting breast cancer cell survival [99].
Acetyl-CoA, produced by acetyl-CoA synthetase 2 (ACSS2) through the catalysis of acetate, is crucial for tumor growth and survival [100, 101]. OGT has been shown to regulate glioblastoma acetate metabolism by influencing cyclin-dependent kinase 5 (CDK5)-dependent ACSS2 phosphorylation. Moreover, drugs targeting OGT and CDK5 have demonstrated efficacy in reducing glioblastoma tumors in vitro [102]. Therefore, OGT plays a significant role in the metabolic reprogramming of tumors and represents a promising therapeutic target (Table 1).
Roles of OGT in drug resistance
Due to the high morbidity and mortality associated with tumors, significant efforts have been made to develop anticancer drugs. These drugs play a crucial role in inhibiting tumor cell metastasis and reducing tumor cell survival. However, tumor cells can alter multiple molecular pathways to develop drug resistance [103, 104].
Platinum-based drugs are commonly used in the treatment of ovarian cancer (OC), but the development of drug resistance remains a significant challenge [105]. Reducing OGT-mediated O-GlcNAcylation of synaptosome-associated protein-23 (SNAP-23) promotes cisplatin resistance by inducing exosome secretion in OC [106]. Additionally, the downregulation of OGT, leading to reduced O-GlcNAcylation of synaptosome-associated protein-29 (SNAP-29), enhances cisplatin-induced autophagy, making OC cells less responsive to cisplatin treatment [107]. Huang et al. demonstrated that OGT interacts with kelch-like ECH-associated protein 1 (KEAP1) and promotes its glycosylation in A2780 and A2780/DDP cell lines. Furthermore, miR-181d enhances OC resistance to cisplatin by regulating the OGT/KEAP1/Nrf2 axis both in vitro and in vivo [108].
5-fluorouracil (5-FU) is a crucial drug for treating colorectal cancer, targeting thymidylate synthase (TS) and its metabolites [109, 110]. Very et al. showed that TS undergoes O-GlcNAcylation through its interaction with OGT, which impedes proteasomal degradation and enhances its stability. Knockdown of OGT reduced cancer cell sensitivity to 5-FU by lowering both TS protein levels and activity [111].
Proteasome inhibitors are used to treat multiple myeloma and mantle cell lymphoma [112]. Inhibition of OGT in NCI-H460 cells and their xenograft model increases cancer cell sensitivity to proteasome inhibitors. This effect is due to the stabilization of nuclear factor erythroid 2-related factor 1 (NRF1) via OGT-catalyzed O-GlcNAcylation, leading to the upregulation of proteasome subunit genes [113].
Osteosarcoma, a common primary bone tumor, shows a reduced survival rate post-metastasis [114]. Methotrexate is an approved therapeutic drug for osteosarcoma [115]. Sun et al. identified that lncRNA EBLN3P increases the resistance of osteosarcoma cells to methotrexate by enhancing the miR-200a-3p/OGT axis [116]. Additionally, docetaxel is a chemotherapy drug approved for prostate cancer treatment [117]. Xia et al. confirmed that miR-140 induces prostate cancer cell sensitivity to docetaxel in an OGT-dependent manner. Knockdown of OGT sensitizes prostate cancer cells to docetaxel [118]. Cisplatin-based systemic chemotherapy is the standard treatment for advanced bladder cancer [119]. Wang et al. observed that reducing OGT expression increased bladder cancer cell sensitivity to cisplatin [120]. Furthermore, gemcitabine and paclitaxel are also used in bladder cancer treatment [121]. OGT knockdown significantly enhanced the sensitivity of drug-resistant bladder cancer cells to these chemotherapy drugs [122].
Based on these studies, OGT-mediated O-GlcNAcylation is associated with tumor treatment resistance. However, current research on targeting OGT for overcoming drug resistance is still limited. This aspect should be further explored in future studies.
Roles of OGT in apoptosis
Apoptosis, first defined as a programmed cell death mode in 1972, is characterized by nuclear and chromatin condensation, as well as the formation of apoptotic bodies, which can be observed through light microscopy [123,124,125]. This process is crucial for maintaining homeostasis in normal tissues, including the gastrointestinal tract, immune system, and skin [126, 127]. However, abnormal apoptosis occurs during tumor progression, leading to reduced apoptosis in tumor cells and enhanced survival [128]. Given OGT’s role in promoting tumor progression, studies have shown that it inhibits tumor cell apoptosis. For instance, in HCC, OGT highly O-GlcNAcylates speckle-type POZ protein (SPOP) at Ser96, facilitating its nuclear entry and inhibiting apoptosis of liver cancer cells [129]. Yu et al. discovered that OGT-mediated O-GlcNAcylation influences integrin α5 (ITGA5) protein stability, promoting tumor cell growth and tumorigenesis while reducing apoptosis [130]. Additionally, OGT O-GlcNAcylates Bmi-1 at Ser255, thus inhibiting apoptosis in prostate cancer cells [131]. Nuclear and spindle-associated protein 1 (NUSAP1) has been shown to promote bladder cancer progression through the TGF-β signaling pathway, and its expression is also associated with lymph node metastasis and survival prognosis [132, 133]. The protein stability of NUSAP1 decreases after knocking down OGT expression in HT-1376 and T24 cells to reduce O-GlcNAcylation, thus promoting bladder cancer cell apoptosis [134]. Therefore, targeting OGT may be advantageous in inducing tumor cell apoptosis (Fig. 3).

Roles of OGT in the regulation of apoptosis, cancer stem-like cells properties, ferroptosis and immune escape in cancer
Roles of OGT in cancer stem-like cells properties
Tumor initiation and progression are regulated by cancer stem cells (CSCs), which possess self-renewal, plasticity, and differentiation capabilities that promote metastasis, drug resistance, and recurrence [135, 136]. Eukaryotic initiation factor 4E (eIF4E) and RAF proto-oncogene serine/threonine-protein kinase (RAF1) are key targets of sorafenib [137, 138]. Research has shown that OGT enhances the stem cell-like potential of HCC cells by upregulating eIF4E [139]. Additionally, OGT establishes a feedback loop with Krüppel-like factor 8 (KLF8), modulating CSC phenotypes and increasing paclitaxel resistance [140]. Therefore, identifying the signaling networks that regulate CSC properties via OGT could contribute to more effective tumor treatments (Fig. 3).
Roles of OGT in ferroptosis
Ferroptosis is a recently identified mode of cell death with unique properties and functions, implicated in various diseases, including tumors, renal disease, and cardiovascular disease [141,142,143]. It is primarily characterized by cytological changes such as the reduction or disappearance of mitochondrial cristae and the condensation of mitochondrial membranes [144, 145]. Recent studies have shown that OGT is associated with ferroptosis in tumors.
Solute carrier family 7, member 11 (SLC7A11), a cystine/glutamate antiporter, promotes cystine import into cells, thereby inhibiting lipid peroxidation and ferroptosis [146, 147]. It has been reported that OGT promotes cystine uptake by HCC cells through O-GlcNAcylation of SLC7A11 at the Ser26 site, leading to ferroptosis inhibition in HCC [148]. Hypoxia-inducible factor 2α (HIF-2α), a hypoxia-related transcription factor, promotes renal cancer progression [149]. OGT was found to increase HIF-2α protein levels in clear cell renal cell carcinoma by inhibiting ubiquitin-proteasome-mediated degradation. Moreover, the OGT/HIF-2α axis modulates the sensitivity of clear cell renal cell carcinoma to ferroptosis [150]. These findings indicate that OGT plays a crucial role in the ferroptosis process, although the regulatory mechanism requires further investigation (Fig. 3).
Roles of OGT in autophagy
Autophagy is a self-degradative process essential for maintaining cellular homeostasis under stress conditions [151, 152]. Well-known regulatory pathways of autophagy include AMPK, PI3K/Akt/mTOR, and Beclin-1 [153]. Recent studies have highlighted the role of OGT in autophagy. Jin et al. demonstrated that overexpression of OGT in bladder cancer cells increases the O-GlcNAcylation level of AMPKα, resulting in altered autophagy flux [154]. Further research is required to elucidate the interaction between OGT and autophagy-related molecules and to characterize its role in tumor progression.
Roles of OGT in immune escape
Since the early 20th century, researchers have explored the role of the immune system in tumor development. Tumor immunosurveillance is a critical process where the immune system monitors, identifies, and eliminates tumor cells [155, 156]. Immune checkpoint proteins, such as Programmed Death-Ligand 1 (PD-L1) and its receptor PD-1, are closely related to tumor immune evasion. PD-L1 interacts with PD-1 on cytotoxic T lymphocytes, transmitting inhibitory signals that weaken the tumor-killing function of these cells [157, 158]. Recent studies have reported that OGT is involved in regulating the expression of immune checkpoint proteins. OGT has been implicated in promoting tumor immune evasion by inhibiting the lysosomal degradation of PD-L1 [159]. Yuan et al. demonstrated that exosomal OGT enhances immune evasion of esophageal cancer stem cells by upregulating PD-1 expression in CD8+ T cells [160]. These findings indicate that OGT plays a role in tumor immune evasion. Further research is needed to determine whether OGT can regulate other immune checkpoint molecules (Fig. 3).
Targeting OGT and O-GlcNAcylation
In most human tumors, OGT functions as an oncoprotein, promoting tumor growth, metastasis, and drug resistance by activating signaling pathways such as proliferation, EMT, and anti-apoptosis. Therefore, targeting OGT is a promising strategy for cancer treatment. For instance, quercetin has been reported to induce cell death in cervical cancer by reducing the expression of OGT, overall O-GlcNAc, and O-GlcNAcylated AMPK [161]. Similarly, corosolic acid inhibits liver cancer progression by decreasing OGT expression and O-GlcNAcylation levels in cancer cells [162]. A number of small molecule compounds targeting OGT activity or OGT-mediated O-GlcNAcylation have been produced, such as OSMI-1 and OSMI-4, and have been widely used in tumor research [163, 164]. OSMI-1 significantly inhibits the proliferation and migration of thyroid cancer cells and slows the occurrence of liver tumors [62, 69].
Moreover, combination therapies have proven more effective than monotherapies in treating tumors. For example, astragalus polysaccharide reduces OGT levels and increases OGA levels in liver cancer cells, thereby downregulating O-GlcNAcylation and promoting doxorubicin-induced apoptosis [165]. A notable increase in cell death has been observed with the coadministration of OSMI-1 and temozolomide. These findings highlight OGT as a promising drug target. However, many phytochemicals and small molecule inhibitors face challenges such as low bioavailability and solubility in human applications. Future applications could overcome these limitations through the use of nanomaterials [166].
Conclusion and perspectives
This review provides an overview of the biological functions of OGT, with a particular focus on its impact on tumors. Elevated levels of OGT expression are commonly observed in tumors. OGT-mediated O-GlcNAcylation promotes tumor cell proliferation and induces EMT, facilitating MMP expression, which is associated with tumor invasion and metastasis. Tumor metabolic reprogramming is also linked to OGT, thereby influencing tumor progression. Furthermore, the OGT/O-GlcNAcylation pathway inhibits apoptosis and ferroptosis, promotes tumor immune escape, and ultimately enhances tumor growth. Notably, OGT mediates drug resistance, making it a critical target for altering tumor cell sensitivity to anticancer treatments. Consequently, small molecule compounds have been developed to inhibit the OGT/O-GlcNAcylation pathway. Knockdown of OGT has been shown to reduce malignant tumor behavior both in vitro and in vivo. Future efforts should focus on translating these research findings into clinical applications to improve cancer patient outcomes.
Data availability
Not applicable.
References
Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21(12):729–49. https://doi.org/10.1038/s41580-020-00294-x.
Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259(5):3308–17.
Holt GD, Snow CM, Senior A, Haltiwanger RS, Gerace L, Hart GW. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. J Cell Biol. 1987;104(5):1157–64. https://doi.org/10.1083/jcb.104.5.1157.
Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Sci (New York NY). 2001;291(5512):2376–8. https://doi.org/10.1126/science.1058714.
Drougat L, Olivier-Van Stichelen S, Mortuaire M, Foulquier F, Lacoste AS, Michalski JC, Lefebvre T, Vercoutter-Edouart AS. Characterization of O-GlcNAc cycling and proteomic identification of differentially O-GlcNAcylated proteins during G1/S transition. Biochim Biophys Acta. 2012;1820(12):1839–48. https://doi.org/10.1016/j.bbagen.2012.08.024.
Liu AR, Ramakrishnan P. Regulation of Nuclear factor-kappab function by O-GlcNAcylation in inflammation and Cancer. Front cell Dev Biology. 2021;9:751761. https://doi.org/10.3389/fcell.2021.751761.
Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, Vocadlo DJ, Seagroves TN, Reginato MJ. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54(5):820–31. https://doi.org/10.1016/j.molcel.2014.04.026.
Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266(8):4706–12.
Akella NM, Ciraku L, Reginato MJ. Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019;17(1):52. https://doi.org/10.1186/s12915-019-0671-3.
Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446(7139):1017–22. https://doi.org/10.1038/nature05815.
Nolte D, Müller U. Human O-GlcNAc transferase (OGT): genomic structure, analysis of splice variants, fine mapping in Xq13.1. Mammalian genome. Official J Int Mammalian Genome Soc. 2002;13(1):62–4. https://doi.org/10.1007/s00335-001-2108-9.
Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature. 2011;469(7331):564–7. https://doi.org/10.1038/nature09638.
Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem. 2000;275(15):10983–8. https://doi.org/10.1074/jbc.275.15.10983.
Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–55. https://doi.org/10.1146/annurev.biochem.76.061005.092322.
Zhang N, Jiang H, Zhang K, Zhu J, Wang Z, Long Y, He Y, Feng F, Liu W, Ye F, Qu W. OGT as potential novel target: structure, function and inhibitors. Chemico-Biol Interact. 2022;357:109886. https://doi.org/10.1016/j.cbi.2022.109886.
Lubas WA, Frank DW, Krause M, Hanover JA. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272(14):9316–24. https://doi.org/10.1074/jbc.272.14.9316.
Hanover JA, Yu S, Lubas WB, Shin SH, Ragano-Caracciola M, Kochran J, Love DC. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch Biochem Biophys. 2003;409(2):287–97. https://doi.org/10.1016/s0003-9861(02)00578-7.
Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272(14):9308–15. https://doi.org/10.1074/jbc.272.14.9308.
Love DC, Kochan J, Cathey RL, Shin SH, Hanover JA. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci. 2003;116(Pt 4):647–54. https://doi.org/10.1242/jcs.00246.
Liu L, Li L, Ma C, Shi Y, Liu C, Xiao Z, Zhang Y, Tian F, Gao Y, Zhang J, Ying W, Wang PG, Zhang L. O-GlcNAcylation of Thr(12)/Ser(56) in short-form O-GlcNAc transferase (sOGT) regulates its substrate selectivity. J Biol Chem. 2019;294(45):16620–33. https://doi.org/10.1074/jbc.RA119.009085.
Allan RK, Ratajczak T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones. 2011;16(4):353–67. https://doi.org/10.1007/s12192-010-0248-0.
Joiner CM, Levine ZG, Aonbangkhen C, Woo CM, Walker S. Aspartate residues far from the active site drive O-GlcNAc transferase substrate selection. J Am Chem Soc. 2019;141(33):12974–8. https://doi.org/10.1021/jacs.9b06061.
Jínek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat Struct Mol Biol. 2004;11(10):1001–7. https://doi.org/10.1038/nsmb833.
Blatch GL, Lässle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. BioEssays: News Reviews Mol Cell Dev Biology. 1999;21(11):932–9. https://doi.org/10.1002/(SICI)1521-1878(199911)21:11%3C932::AID-BIES5%3E3.0.CO;2-N.
Wang Y, Shu H, Liu J, Jin X, Wang L, Qu Y, Xia M, Peng P, Feng Y, Wei M. EGF promotes PKM2 O-GlcNAcylation by stimulating O-GlcNAc transferase phosphorylation at Y976 and their subsequent association. J Biol Chem. 2022;298(9):102340. https://doi.org/10.1016/j.jbc.2022.102340.
Ding X, Jiang W, Zhou P, Liu L, Wan X, Yuan X, Wang X, Chen M, Chen J, Yang J, Kong C, Li B, Peng C, Wong CC, Hou F, Zhang Y. Mixed lineage leukemia 5 (MLL5) Protein Stability is cooperatively regulated by O-GlcNac Transferase (OGT) and Ubiquitin specific protease 7 (USP7). PLoS ONE. 2015;10(12):e0145023. https://doi.org/10.1371/journal.pone.0145023.
Zhai L, Yang X, Dong J, Qian L, Gao Y, Lv Y, Chen L, Chen B, Zhou F. O–GlcNAcylation mediates endometrial cancer progression by regulating the Hippo–YAP pathway. Int J Oncol. 2023;63(2). https://doi.org/10.3892/ijo.2023.5538.
Wang Y, Wang G, Liu Y, Yang F, Zhang H, Kong Y. Icaritin inhibits endometrial carcinoma cells by suppressing O-GlcNAcylation of FOXC1. Phytomedicine: Int J Phytotherapy Phytopharmacology. 2023;120:155062. https://doi.org/10.1016/j.phymed.2023.155062.
Poulose N, Forsythe N, Polonski A, Gregg G, Maguire S, Fuchs M, Minner S, Sauter G, McDade SS, Mills IG. VPRBP functions downstream of the androgen receptor and OGT to restrict p53 activation in prostate Cancer. Mol cancer Research: MCR. 2022;20(7):1047–60. https://doi.org/10.1158/1541-7786.Mcr-21-0477.
Shen H, Zhao X, Chen J, Qu W, Huang X, Wang M, Shao Z, Shu Q, Li X. O-GlcNAc transferase ogt regulates embryonic neuronal development through modulating Wnt/β-catenin signaling. Hum Mol Genet. 2021;31(1):57–68. https://doi.org/10.1093/hmg/ddab223.
Levine ZG, Potter SC, Joiner CM, Fei GQ, Nabet B, Sonnett M, Zachara NE, Gray NS, Paulo JA, Walker S. Mammalian cell proliferation requires noncatalytic functions of O-GlcNAc transferase. Proc Natl Acad Sci USA. 2021;118(4). https://doi.org/10.1073/pnas.2016778118.
Li X, Yue X, Sepulveda H, Burt RA, Scott DA, S, AC SAM, Rao A. OGT controls mammalian cell viability by regulating the proteasome/mTOR/ mitochondrial axis. Proc Natl Acad Sci USA. 2023;120(3):e2218332120. https://doi.org/10.1073/pnas.2218332120.
Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain. 2009;132(Pt 7):1820–32. https://doi.org/10.1093/brain/awp099.
Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, Zhu G, Zhao Y, Chen Y, Yu Y, Pan Q, Wang J, Sun F. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun. 2017;8:15280. https://doi.org/10.1038/ncomms15280.
Jiang M, Qiu Z, Zhang S, Fan X, Cai X, Xu B, Li X, Zhou J, Zhang X, Chu Y, Wang W, Liang J, Horvath T, Yang X, Wu K, Nie Y, Fan D. Elevated O-GlcNAcylation promotes gastric cancer cells proliferation by modulating cell cycle related proteins and ERK 1/2 signaling. Oncotarget. 2016;7(38):61390–402. https://doi.org/10.18632/oncotarget.11359.
Zhu G, Qian M, Lu L, Chen Y, Zhang X, Wu Q, Liu Y, Bian Z, Yang Y, Guo S, Wang J, Pan Q, Sun F. O-GlcNAcylation of YY1 stimulates tumorigenesis in colorectal cancer cells by targeting SLC22A15 and AANAT. Carcinogenesis. 2019;40(9):1121–31. https://doi.org/10.1093/carcin/bgz010.
Shi R, Tao J, Jiang X, Li M, Zhu R, Luo S, Lu Z. Fructose-1,6-bisphosphatase 1 suppresses PPARα-mediated gene transcription and non-small-cell lung cancer progression. Am J cancer Res. 2023;13(10):4742–54.
Wang Z, Li M, Jiang H, Luo S, Shao F, Xia Y, Yang M, Ren X, Liu T, Yan M, Qian X, He H, Guo D, Duan Y, Wu K, Wang L, Ji G, Shen Y, Li L, Zheng P, Dong B, Fang J, Zheng M, Liang T, Li H, Yu R, Xu D, Lu Z. Fructose-1,6-bisphosphatase 1 functions as a protein phosphatase to dephosphorylate histone H3 and suppresses PPARα-regulated gene transcription and tumour growth. Nat Cell Biol. 2022;24(11):1655–65. https://doi.org/10.1038/s41556-022-01009-4.
Yin Q, Zheng M, Luo Q, Jiang D, Zhang H, Chen C. YB-1 as an oncoprotein: functions, Regulation, post-translational modifications, and targeted therapy. Cells. 2022;11(7). https://doi.org/10.3390/cells11071217.
Tao Z, Ruan H, Sun L, Kuang D, Song Y, Wang Q, Wang T, Hao Y, Chen K. Targeting the YB-1/PD-L1 Axis to Enhance Chemotherapy and Antitumor Immunity. Cancer Immunol Res. 2019;7(7):1135–47. https://doi.org/10.1158/2326-6066.Cir-18-0648.
Liu Q, Tao T, Liu F, Ni R, Lu C, Shen A. Hyper-O-GlcNAcylation of YB-1 affects Ser102 phosphorylation and promotes cell proliferation in hepatocellular carcinoma. Exp Cell Res. 2016;349(2):230–8. https://doi.org/10.1016/j.yexcr.2016.10.011.
Zhou P, Chang WY, Gong DA, Xia J, Chen W, Huang LY, Liu R, Liu Y, Chen C, Wang K, Tang N, Huang AL. High dietary fructose promotes hepatocellular carcinoma progression by enhancing O-GlcNAcylation via microbiota-derived acetate. Cell Metabol. 2023;35(11):1961–75. .e6.
Ullah R, Yin Q, Snell AH, Wan L. RAF-MEK-ERK pathway in cancer evolution and treatment. Sem Cancer Biol. 2022;85:123–54. https://doi.org/10.1016/j.semcancer.2021.05.010.
Xu Y, Sheng X, Zhao T, Zhang L, Ruan Y, Lu H. O-GlcNAcylation of MEK2 promotes the proliferation and migration of breast cancer cells. Glycobiology. 2021;31(5):571–81. https://doi.org/10.1093/glycob/cwaa103.
Chai Y, Du Y, Zhang S, Xiao J, Luo Z, He F, Huang K. MicroRNA-485-5p reduces O-GlcNAcylation of Bmi-1 and inhibits colorectal cancer proliferation. Exp Cell Res. 2018;368(1):111–8. https://doi.org/10.1016/j.yexcr.2018.04.020.
Li J, Ahmad M, Sang L, Zhan Y, Wang Y, Yan Y, Liu Y, Mi W, Lu M, Dai Y, Zhang R, Dong MQ, Yang YG, Wang X, Sun J, Li J. O-GlcNAcylation promotes the cytosolic localization of the m(6)a reader YTHDF1 and colorectal cancer tumorigenesis. J Biol Chem. 2023;299(6):104738. https://doi.org/10.1016/j.jbc.2023.104738.
Su Z, Gao A, Li X, Zou S, He C, Wu J, Ding WQ, Zhou J. DNA polymerase Iota promotes esophageal squamous cell Carcinoma Proliferation through Erk-OGT-Induced G6PD overactivation. Front Oncol. 2021;11:706337. https://doi.org/10.3389/fonc.2021.706337.
Yu FY, Zhou CY, Liu YB, Wang B, Mao L, Li Y. miR-483 is down-regulated in gastric cancer and suppresses cell proliferation, invasion and protein O-GlcNAcylation by targeting OGT. Neoplasma. 2018;65(3):406–14. https://doi.org/10.4149/neo_2018_170608N411.
Ning D, Chen J, Du P, Liu Q, Cheng Q, Li X, Zhang B, Chen X, Jiang L. The crosstalk network of XIST/miR-424-5p/OGT mediates RAF1 glycosylation and participates in the progression of liver cancer. Liver International: Official J Int Association Study Liver. 2021;41(8):1933–44. https://doi.org/10.1111/liv.14904.
Zheng W, Li H, Zhang H, Zhang C, Zhu Z, Liang H, Zhou Y. Long noncoding RNA RHPN1-AS1 promotes colorectal cancer progression via targeting miR-7-5p/OGT axis. Cancer Cell Int. 2020;20:54. https://doi.org/10.1186/s12935-020-1110-9.
Gerstberger S, Jiang Q, Ganesh K, Metastasis. Cell. 2023;186(8):1564–79. https://doi.org/10.1016/j.cell.2023.03.003.
Mittal V. Epithelial mesenchymal transition in Tumor Metastasis. Annu Rev Pathol. 2018;13:395–412. https://doi.org/10.1146/annurev-pathol-020117-043854.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84. https://doi.org/10.1038/s41580-018-0080-4.
Jiang M, Xu B, Li X, Shang Y, Chu Y, Wang W, Chen D, Wu N, Hu S, Zhang S, Li M, Wu K, Yang X, Liang J, Nie Y, Fan D. O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene. 2019;38(3):301–16. https://doi.org/10.1038/s41388-018-0435-5.
Ge X, Peng X, Li M, Ji F, Chen J, Zhang D. OGT regulated O-GlcNacylation promotes migration and invasion by activating IL-6/STAT3 signaling in NSCLC cells. Pathol Res Pract. 2021;225:153580. https://doi.org/10.1016/j.prp.2021.153580.
Jin FZ, Yu C, Zhao DZ, Wu MJ, Yang Z. A correlation between altered O-GlcNAcylation, migration and with changes in E-cadherin levels in ovarian cancer cells. Exp Cell Res. 2013;319(10):1482–90. https://doi.org/10.1016/j.yexcr.2013.03.013.
de Almeida LGN, Thode H, Eslambolchi Y, Chopra S, Young D, Gill S, Devel L, Dufour A. Matrix metalloproteinases: from Molecular mechanisms to Physiology, Pathophysiology, and Pharmacology. Pharmacol Rev. 2022;74(3):712–68. https://doi.org/10.1124/pharmrev.121.000349.
Zitka O, Kukacka J, Krizkova S, Huska D, Adam V, Masarik M, Prusa R, Kizek R. Matrix metalloproteinases. Curr Med Chem. 2010;17(31):3751–68. https://doi.org/10.2174/092986710793213724.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. https://doi.org/10.1016/j.cell.2010.03.015.
Qiao Z, Dang C, Zhou B, Li S, Zhang W, Jiang J, Zhang J, Ma Y, Kong R, Ma Z. Downregulation of O-linked N-acetylglucosamine transferase by RNA interference decreases MMP9 expression in human esophageal cancer cells. Oncol Lett. 2016;11(5):3317–23. https://doi.org/10.3892/ol.2016.4428.
Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem. 2012;287(14):11070–81. https://doi.org/10.1074/jbc.M111.302547.
Li X, Wu Z, He J, Jin Y, Chu C, Cao Y, Gu F, Wang H, Hou C, Liu X, Zou Q. OGT regulated O-GlcNAcylation promotes papillary thyroid cancer malignancy via activating YAP. Oncogene. 2021;40(30):4859–71. https://doi.org/10.1038/s41388-021-01901-7.
Lv Z, Ma G, Zhong Z, Xie X, Li B, Long D. O-GlcNAcylation of RAB10 promotes hepatocellular carcinoma progression. Carcinogenesis. 2023;44(10–11):785–94. https://doi.org/10.1093/carcin/bgad034.
Hao JW, Wang J, Guo H, Zhao YY, Sun HH, Li YF, Lai XY, Zhao N, Wang X, Xie C, Hong L, Huang X, Wang HR, Li CB, Liang B, Chen S, Zhao TJ. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat Commun. 2020;11(1):4765. https://doi.org/10.1038/s41467-020-18565-8.
Son NH, Basu D, Samovski D, Pietka TA, Peche VS, Willecke F, Fang X, Yu SQ, Scerbo D, Chang HR, Sun F, Bagdasarov S, Drosatos K, Yeh ST, Mullick AE, Shoghi KI, Gumaste N, Kim K, Huggins LA, Lhakhang T, Abumrad NA, Goldberg IJ. Endothelial cell CD36 optimizes tissue fatty acid uptake. J Clin Investig. 2018;128(10):4329–42. https://doi.org/10.1172/jci99315.
Jiang M, Wu N, Xu B, Chu Y, Li X, Su S, Chen D, Li W, Shi Y, Gao X, Zhang H, Zhang Z, Du W, Nie Y, Liang J, Fan D. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics. 2019;9(18):5359–73. https://doi.org/10.7150/thno.34024.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, He C. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20. https://doi.org/10.1038/nature12730.
Lee Y, Choe J, Park OH, Kim YK. Molecular mechanisms driving mRNA degradation by m(6)a modification. Trends Genet. 2020;36(3):177–88. https://doi.org/10.1016/j.tig.2019.12.007.
Yang Y, Yan Y, Yin J, Tang N, Wang K, Huang L, Hu J, Feng Z, Gao Q, Huang A. O-GlcNAcylation of YTHDF2 promotes HBV-related hepatocellular carcinoma progression in an N(6)-methyladenosine-dependent manner. Signal Transduct Target Therapy. 2023;8(1):63. https://doi.org/10.1038/s41392-023-01316-8.
Wang G, Xu Z, Sun J, Liu B, Ruan Y, Gu J, Song S. O-GlcNAcylation enhances Reticulon 2 protein stability and its promotive effects on gastric cancer progression. Cell Signal. 2023;108:110718. https://doi.org/10.1016/j.cellsig.2023.110718.
Lin CH, Liao CC, Wang SY, Peng CY, Yeh YC, Chen MY, Chou TY. Comparative O-GlcNAc proteomic analysis reveals a role of O-GlcNAcylated SAM68 in Lung Cancer aggressiveness. Cancers. 2022;14(1). https://doi.org/10.3390/cancers14010243.
Liu HY, Liu YY, Yang F, Zhang L, Zhang FL, Hu X, Shao ZM, Li DQ. Acetylation of MORC2 by NAT10 regulates cell-cycle checkpoint control and resistance to DNA-damaging chemotherapy and radiotherapy in breast cancer. Nucleic Acids Res. 2020;48(7):3638–56. https://doi.org/10.1093/nar/gkaa130.
Zhang FL, Yang SY, Liao L, Zhang TM, Zhang YL, Hu SY, Deng L, Huang MY, Andriani L, Ma XY, Shao ZM, Li DQ. Dynamic SUMOylation of MORC2 orchestrates chromatin remodelling and DNA repair in response to DNA damage and drives chemoresistance in breast cancer. Theranostics. 2023;13(3):973–90. https://doi.org/10.7150/thno.79688.
Liu YY, Liu HY, Yu TJ, Lu Q, Zhang FL, Liu GY, Shao ZM, Li DQ. O-GlcNAcylation of MORC2 at threonine 556 by OGT couples TGF-β signaling to breast cancer progression. Cell Death Differ. 2022;29(4):861–73. https://doi.org/10.1038/s41418-021-00901-0.
Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, Reginato MJ. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29(19):2831–42. https://doi.org/10.1038/onc.2010.41.
Liu Y, Huang H, Cao Y, Wu Q, Li W, Zhang J. Suppression of OGT by microRNA24 reduces FOXA1 stability and prevents breast cancer cells invasion. Biochem Biophys Res Commun. 2017;487(3):755–62. https://doi.org/10.1016/j.bbrc.2017.04.135.
Tao T, He Z, Shao Z, Lu H. Table 3 O-GlcNAcylation promotes metastasis of triple negative breast cancer. Oncotarget. 2016;7(16):22807–18. https://doi.org/10.18632/oncotarget.8182.
Mirzaei S, Saghari S, Bassiri F, Raesi R, Zarrabi A, Hushmandi K, Sethi G, Tergaonkar V. NF-κB as a regulator of cancer metastasis and therapy response: a focus on epithelial-mesenchymal transition. J Cell Physiol. 2022;237(7):2770–95. https://doi.org/10.1002/jcp.30759.
Mortezaee K, Najafi M, Farhood B, Ahmadi A, Shabeeb D, Musa AE. NF-κB targeting for overcoming tumor resistance and normal tissues toxicity. J Cell Physiol. 2019;234(10):17187–204. https://doi.org/10.1002/jcp.28504.
Ali A, Kim SH, Kim MJ, Choi MY, Kang SS, Cho GJ, Kim YS, Choi JY, Choi WS. O-GlcNAcylation of NF-κB promotes Lung Metastasis of Cervical Cancer cells via Upregulation of CXCR4 expression. Mol Cells. 2017;40(7):476–84. https://doi.org/10.14348/molcells.2017.2309.
Niu Y, Xia Y, Wang J, Shi X. O-GlcNAcylation promotes migration and invasion in human ovarian cancer cells via the RhoA/ROCK/MLC pathway. Mol Med Rep. 2017;15(4):2083–9. https://doi.org/10.3892/mmr.2017.6244.
Seo HG, Kim HB, Yoon JY, Kweon TH, Park YS, Kang J, Jung J, Son S, Yi EC, Lee TH, Yang WH, Cho JW. Mutual regulation between OGT and XIAP to control colon cancer cell growth and invasion. Cell Death Dis. 2020;11(9):815. https://doi.org/10.1038/s41419-020-02999-5.
Wu N, Jiang M, Han Y, Liu H, Chu Y, Liu H, Cao J, Hou Q, Zhao Y, Xu B, Xie X. O-GlcNAcylation promotes colorectal cancer progression by regulating protein stability and potential catcinogenic function of DDX5. J Cell Mol Med. 2019;23(2):1354–62. https://doi.org/10.1111/jcmm.14038.
Huang X, Pan Q, Sun D, Chen W, Shen A, Huang M, Ding J, Geng M. O-GlcNAcylation of cofilin promotes breast cancer cell invasion. J Biol Chem. 2013;288(51):36418–25. https://doi.org/10.1074/jbc.M113.495713.
Wang L, Li G, Zhou Z, Ge C, Chen Q, Liu Y, Zhang N, Zhang K, Niu M, Li W, Zhong X, Wu S, Zhang J, Liu Y. Chromatin-associated OGT promotes the malignant progression of hepatocellular carcinoma by activating ZNF263. Oncogene. 2023;42(30):2329–46. https://doi.org/10.1038/s41388-023-02751-1.
Zhu G, Tao T, Zhang D, Liu X, Qiu H, Han L, Xu Z, Xiao Y, Cheng C, Shen A. O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology. 2016;26(8):820–33. https://doi.org/10.1093/glycob/cww025.
Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discovery. 2022;21(2):141–62. https://doi.org/10.1038/s41573-021-00339-6.
Yu T, Wang Y, Fan Y, Fang N, Wang T, Xu T, Shu Y. CircRNAs in cancer metabolism: a review. J Hematol Oncol. 2019;12(1):90. https://doi.org/10.1186/s13045-019-0776-8.
Mikawa T, ME LL, Takaori-Kondo A, Inagaki N, Yokode M, Kondoh H. Dysregulated glycolysis as an oncogenic event. Cell Mol Life Sci. 2015;72(10):1881–92. https://doi.org/10.1007/s00018-015-1840-3.
Hu H, Zhu W, Qin J, Chen M, Gong L, Li L, Liu X, Tao Y, Yin H, Zhou H, Zhou L, Ye D, Ye Q, Gao D. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology (Baltimore MD). 2017;65(2):515–28. https://doi.org/10.1002/hep.28887.
He Y, Wang X, Lu W, Zhang D, Huang L, Luo Y, Xiong L, Li H, Zhang P, Li Q, Liang S. PGK1 contributes to tumorigenesis and sorafenib resistance of renal clear cell carcinoma via activating CXCR4/ERK signaling pathway and accelerating glycolysis. Cell Death Dis. 2022;13(2):118. https://doi.org/10.1038/s41419-022-04576-4.
Liang J, Liu C, Xu D, Xie K, Li A. LncRNA NEAT1 facilitates glioma progression via stabilizing PGK1. J Translational Med. 2022;20(1):80. https://doi.org/10.1186/s12967-022-03273-2.
Nie H, Ju H, Fan J, Shi X, Cheng Y, Cang X, Zheng Z, Duan X, Yi W. O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat Commun. 2020;11(1):36. https://doi.org/10.1038/s41467-019-13601-8.
Singh JP, Qian K, Lee JS, Zhou J, Han X, Zhang B, Ong Q, Ni W, Jiang M, Ruan HB, Li MD, Zhang K, Ding Z, Lee P, Singh K, Wu J, Herzog RI, Kaech S, Wendel HG, Yates JR 3rd, Han W, Sherwin RS, Nie Y, Yang X. O-GlcNAcase targets pyruvate kinase M2 to regulate tumor growth. Oncogene. 2020;39(3):560–73. https://doi.org/10.1038/s41388-019-0975-3.
He X, Wu N, Li R, Zhang H, Zhao Y, Nie Y, Wu J. IDH2, a novel target of OGT, facilitates glucose uptake and cellular bioenergy production via NF-κB signaling to promote colorectal cancer progression. Cell Oncol (Dordrecht). 2023;46(1):145–64. https://doi.org/10.1007/s13402-022-00740-2.
Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021;218(1). https://doi.org/10.1084/jem.20201606.
Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and Furious: lipid metabolism in Antitumoral Therapy Response and Resistance. Trends cancer. 2021;7(3):198–213. https://doi.org/10.1016/j.trecan.2020.10.004.
Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Design. 2014;20(15):2619–26. https://doi.org/10.2174/13816128113199990486.
Sodi VL, Bacigalupa ZA, Ferrer CM, Lee JV, Gocal WA, Mukhopadhyay D, Wellen KE, Ivan M, Reginato MJ. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene. 2018;37(7):924–34. https://doi.org/10.1038/onc.2017.395.
Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16(11):708–17. https://doi.org/10.1038/nrc.2016.87.
Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159(7):1603–14. https://doi.org/10.1016/j.cell.2014.11.025.
Ciraku L, Bacigalupa ZA, Ju J, Moeller RA, Le Minh G, Lee RH, Smith MD, Ferrer CM, Trefely S, Izzo LT, Doan MT, Gocal WA, D’Agostino L, Shi W, Jackson JG, Katsetos CD, Wellen KE, Snyder NW, Reginato MJ. O-GlcNAc transferase regulates glioblastoma acetate metabolism via regulation of CDK5-dependent ACSS2 phosphorylation. Oncogene. 2022;41(14):2122–36. https://doi.org/10.1038/s41388-022-02237-6.
Su KJ, Yu YL. Downregulation of SHIP2 by Hepatitis B Virus X promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2. Cancers. 2019;11(8). https://doi.org/10.3390/cancers11081065.
Zhang H, Wang Q, Liu J, Cao H. Inhibition of the PI3K/Akt signaling pathway reverses sorafenib-derived chemo-resistance in hepatocellular carcinoma. Oncol Lett. 2018;15(6):9377–84. https://doi.org/10.3892/ol.2018.8536.
Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Reviews Disease Primers. 2016;2:16061. https://doi.org/10.1038/nrdp.2016.61.
Qian L, Yang X, Li S, Zhao H, Gao Y, Zhao S, Lv X, Zhang X, Li L, Zhai L, Zhou F, Chen B. Reduced O-GlcNAcylation of SNAP-23 promotes cisplatin resistance by inducing exosome secretion in ovarian cancer. Cell Death Discovery. 2021;7(1):112. https://doi.org/10.1038/s41420-021-00489-x.
Zhou F, Yang X, Zhao H, Liu Y, Feng Y, An R, Lv X, Li J, Chen B. Down-regulation of OGT promotes cisplatin resistance by inducing autophagy in ovarian cancer. Theranostics. 2018;8(19):5200–12. https://doi.org/10.7150/thno.27806.
Huang W, Chen L, Zhu K, Wang D. Oncogenic microRNA-181d binding to OGT contributes to resistance of ovarian cancer cells to cisplatin. Cell Death Discovery. 2021;7(1):379. https://doi.org/10.1038/s41420-021-00715-6.
Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206:107447. https://doi.org/10.1016/j.pharmthera.2019.107447.
Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8. https://doi.org/10.1038/nrc1074.
Very N, Hardivillé S, Decourcelle A, Thévenet J, Djouina M, Page A, Vergoten G, Schulz C, Kerr-Conte J, Lefebvre T, Dehennaut V. El Yazidi-Belkoura I. Thymidylate synthase O-GlcNAcylation: a molecular mechanism of 5-FU sensitization in colorectal cancer. Oncogene. 2022;41(5):745–56. https://doi.org/10.1038/s41388-021-02121-9.
Fricker LD. Proteasome inhibitor drugs. Annu Rev Pharmacol Toxicol. 2020;60:457–76. https://doi.org/10.1146/annurev-pharmtox-010919-023603.
Sekine H, Okazaki K, Kato K, Alam MM, Shima H, Katsuoka F, Tsujita T, Suzuki N, Kobayashi A, Igarashi K, Yamamoto M, Motohashi H. O-GlcNAcylation Signal mediates proteasome inhibitor resistance in Cancer cells by stabilizing NRF1. Mol Cell Biol. 2018;38(17). https://doi.org/10.1128/mcb.00252-18.
Shoaib Z, Fan TM, Irudayaraj JMK. Osteosarcoma mechanobiology and therapeutic targets. Br J Pharmacol. 2022;179(2):201–17. https://doi.org/10.1111/bph.15713.
Hattinger CM, Tavanti E, Fanelli M, Vella S, Picci P, Serra M. Pharmacogenomics of genes involved in antifolate drug response and toxicity in osteosarcoma. Expert Opin Drug Metab Toxicol. 2017;13(3):245–57. https://doi.org/10.1080/17425255.2017.1246532.
Sun MX, An HY, Sun YB, Sun YB, Bai B. LncRNA EBLN3P attributes methotrexate resistance in osteosarcoma cells through miR-200a-3p/O-GlcNAc transferase pathway. J Orthop Surg Res. 2022;17(1):557. https://doi.org/10.1186/s13018-022-03449-y.
Schaeffer EM, Srinivas S, Adra N, An Y, Barocas D, Bitting R, Bryce A, Chapin B, Cheng HH, D’Amico AV, Desai N, Dorff T, Eastham JA, Farrington TA, Gao X, Gupta S, Guzzo T, Ippolito JE, Kuettel MR, Lang JM, Lotan T, McKay RR, Morgan T, Netto G, Pow-Sang JM, Reiter R, Roach M, Robin T, Rosenfeld S, Shabsigh A, Spratt D, Teply BA, Tward J, Valicenti R, Wong JK, Shead DA, Snedeker J, Freedman-Cass DA. Prostate Cancer, Version 4.2023, NCCN Clinical Practice guidelines in Oncology. J Natl Compr Cancer Network: JNCCN. 2023;21(10):1067–96. https://doi.org/10.6004/jnccn.2023.0050.
Xia M, Wang S, Qi Y, Long K, Li E, He L, Pan F, Guo Z, Hu Z. Inhibition of O-GlcNAc transferase sensitizes prostate cancer cells to docetaxel. Front Oncol. 2022;12:993243. https://doi.org/10.3389/fonc.2022.993243.
Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström PU, Choi W, Guo CC, Lotan Y, Kassouf W. Bladder cancer. Lancet (London England). 2016;388(10061):2796–810. https://doi.org/10.1016/s0140-6736(16)30512-8.
Wang L, Chen S, Zhang Z, Zhang J, Mao S, Zheng J, Xuan Y, Liu M, Cai K, Zhang W, Guo Y, Zhai W, Yao X. Suppressed OGT expression inhibits cell proliferation while inducing cell apoptosis in bladder cancer. BMC Cancer. 2018;18(1):1141. https://doi.org/10.1186/s12885-018-5033-y.
Calabrò F, Lorusso V, Rosati G, Manzione L, Frassineti L, Sava T, Di Paula ED, Alonso S, Sternberg CN. Gemcitabine and paclitaxel every 2 weeks in patients with previously untreated urothelial carcinoma. Cancer. 2009;115(12):2652–9. https://doi.org/10.1002/cncr.24313.
Lee HW, Kang MJ, Kwon YJ, Abdi Nansa S, Jung EH, Kim SH, Lee SJ, Jeong KC, Kim Y, Cheong H, Seo HK. Targeted inhibition of O-Linked β-N-Acetylglucosamine transferase as a Promising Therapeutic Strategy to Restore Chemosensitivity and Attenuate Aggressive Tumor traits in Chemoresistant Urothelial Carcinoma of the bladder. Biomedicines. 2022;10(5). https://doi.org/10.3390/biomedicines10051162.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. https://doi.org/10.1038/bjc.1972.33.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. https://doi.org/10.1080/01926230701320337.
Corcoran GB, Fix L, Jones DP, Moslen MT, Nicotera P, Oberhammer FA, Buttyan R. Apoptosis: molecular control point in toxicity. Toxicol Appl Pharmcol. 1994;128(2):169–81. https://doi.org/10.1006/taap.1994.1195.
Bold RJ, Termuhlen PM, McConkey DJ. Apoptosis, cancer and cancer therapy. Surg Oncol. 1997;6(3):133–42. https://doi.org/10.1016/s0960-7404(97)00015-7.
Evan GI, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7(6):825–34. https://doi.org/10.1016/0955-0674(95)80066-2.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Experimental Clin cancer Research: CR. 2011;30(1):87. https://doi.org/10.1186/1756-9966-30-87.
Zhou P, Chang WY, Gong DA, Huang LY, Liu R, Liu Y, Xia J, Wang K, Tang N, Huang AL. O-GlcNAcylation of SPOP promotes carcinogenesis in hepatocellular carcinoma. Oncogene. 2023;42(10):725–36. https://doi.org/10.1038/s41388-022-02589-z.
Yu M, Chu S, Fei B, Fang X, Liu Z. O-GlcNAcylation of ITGA5 facilitates the occurrence and development of colorectal cancer. Exp Cell Res. 2019;382(2):111464. https://doi.org/10.1016/j.yexcr.2019.06.009.
Li Y, Wang L, Liu J, Zhang P, An M, Han C, Li Y, Guan X, Zhang K. O-GlcNAcylation modulates Bmi-1 protein stability and potential oncogenic function in prostate cancer. Oncogene. 2017;36(45):6293–305. https://doi.org/10.1038/onc.2017.223.
Gao S, Yin H, Tong H, Zhan K, Yang G, Hossain MA, Li T, Gou X, He W. Nucleolar and Spindle Associated Protein 1 (NUSAP1) promotes bladder Cancer Progression through the TGF-β signaling pathway. OncoTargets Therapy. 2020;13:813–25. https://doi.org/10.2147/ott.S237127.
Hou J, Lu Z, Liu X, Luo B, Qu G, Xu Y, Tang C. Increased NUSAP1 expression is associated with lymph node metastasis and survival prognosis in bladder urothelial carcinoma. Sci Rep. 2022;12(1):7003. https://doi.org/10.1038/s41598-022-11137-4.
Chen Y, Liu J, Zhang W, Kadier A, Wang R, Zhang H, Yao X. O-GlcNAcylation enhances NUSAP1 Stability and promotes bladder Cancer aggressiveness. OncoTargets Therapy. 2021;14:445–54. https://doi.org/10.2147/ott.S258175.
Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019;234:116781. https://doi.org/10.1016/j.lfs.2019.116781.
Kharkar PS. Cancer stem cell (CSC) inhibitors: a review of recent patents (2012–2015). Expert Opin Ther Pat. 2017;27(7):753–61. https://doi.org/10.1080/13543776.2017.1325465.
Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol Research: Official J Japan Soc Hepatol. 2013;43(2):147–54. https://doi.org/10.1111/j.1872-034X.2012.01113.x.
Yuan J, Lv T, Yang J, Wu Z, Yan L, Yang J, Shi Y, Jiang L. HDLBP promotes Hepatocellular Carcinoma Proliferation and Sorafenib Resistance by suppressing Trim71-dependent RAF1 degradation. Cell Mol Gastroenterol Hepatol. 2023;15(2):307–25. https://doi.org/10.1016/j.jcmgh.2022.10.005.
Cao B, Duan M, Xing Y, Liu C, Yang F, Li Y, Yang T, Wei Y, Gao Q, Jiang J. O-GlcNAc transferase activates stem-like cell potential in hepatocarcinoma through O-GlcNAcylation of eukaryotic initiation factor 4E. J Cell Mol Med. 2019;23(4):2384–98. https://doi.org/10.1111/jcmm.14043.
Le Minh G, Esquea EM, Dhameliya TT, Merzy J, Lee MH, Ball LE, Reginato MJ. Kruppel-like factor 8 regulates triple negative breast cancer stem cell-like activity. Front Oncol. 2023;13:1141834. https://doi.org/10.3389/fonc.2023.1141834.
Liang C, Zhang X, Yang M, Dong X. Recent progress in Ferroptosis Inducers for Cancer Therapy. Advanced materials (Deerfield Beach. Fla). 2019;31(51):e1904197. https://doi.org/10.1002/adma.201904197.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82. https://doi.org/10.1038/s41580-020-00324-8.
Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11(7):3052–9. https://doi.org/10.7150/thno.54113.
Latunde-Dada GO, Ferroptosis. Role of lipid peroxidation, iron and ferritinophagy. Biochimica et biophysica acta General subjects. 2017;1861(8):1893–900. https://doi.org/10.1016/j.bbagen.2017.05.019
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79. https://doi.org/10.1038/cdd.2015.158.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599–620. https://doi.org/10.1007/s13238-020-00789-5.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, Li J, Li W, Wei S, Vatan L, Zhang H, Szeliga W, Gu W, Liu R, Lawrence TS, Lamb C, Tanno Y, Cieslik M, Stone E, Georgiou G, Chan TA, Chinnaiyan A, Zou W. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4. https://doi.org/10.1038/s41586-019-1170-y.
Tang J, Long G, Hu K, Xiao D, Liu S, Xiao L, Zhou L, Tao Y. Targeting USP8 inhibits O-GlcNAcylation of SLC7A11 to promote Ferroptosis of Hepatocellular Carcinoma via stabilization of OGT. Adv Sci (Weinheim Baden-Wurttemberg Germany). 2023;10(33):e2302953. https://doi.org/10.1002/advs.202302953.
Schödel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, Mole DR, Hypoxia. Hypoxia-inducible transcription factors, and Renal Cancer. Eur Urol. 2016;69(4):646–57. https://doi.org/10.1016/j.eururo.2015.08.007.
Yang Z, Wei X, Ji C, Ren X, Su W, Wang Y, Zhou J, Zhao Z, Zhou P, Zhao K, Yao B, Song N, Qin C. OGT/HIF-2α axis promotes the progression of clear cell renal cell carcinoma and regulates its sensitivity to ferroptosis. iScience. 2023;26(11):108148. https://doi.org/10.1016/j.isci.2023.108148.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. https://doi.org/10.1002/path.2697.
Liu S, Yao S, Yang H, Liu S, Wang Y, Autophagy. Regulator of cell death. Cell Death Dis. 2023;14(10):648. https://doi.org/10.1038/s41419-023-06154-8.
Ganzleben I, Neurath MF, Becker C. Autophagy in Cancer Therapy-Molecular mechanisms and current clinical advances. Cancers. 2021;13(21). https://doi.org/10.3390/cancers13215575.
Jin L, Yuan F, Dai G, Yao Q, Xiang H, Wang L, Xue B, Shan Y, Liu X. Blockage of O-linked GlcNAcylation induces AMPK-dependent autophagy in bladder cancer cells. Cell Mol Biol Lett. 2020;25:17. https://doi.org/10.1186/s11658-020-00208-x.
Kostecki KL, Iida M, Crossman BE, Salgia R, Harari PM, Bruce JY, Wheeler DL. Immune escape strategies in Head and Neck Cancer: evade, resist, inhibit, Recruit. Cancers. 2024;16(2). https://doi.org/10.3390/cancers16020312.
Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of immune evasion in breast cancer. BMC Cancer. 2018;18(1):556. https://doi.org/10.1186/s12885-018-4441-3.
Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 expression in Cancer. Mol Cell. 2019;76(3):359–70. https://doi.org/10.1016/j.molcel.2019.09.030.
Gaikwad S, Agrawal MY, Kaushik I, Ramachandran S, Srivastava SK. Immune checkpoint proteins: signaling mechanisms and molecular interactions in cancer immunotherapy. Sem Cancer Biol. 2022;86(Pt 3):137–50. https://doi.org/10.1016/j.semcancer.2022.03.014.
Zhu Q, Wang H, Chai S, Xu L, Lin B, Yi W, Wu L. O-GlcNAcylation promotes tumor immune evasion by inhibiting PD-L1 lysosomal degradation. Proc Natl Acad Sci USA. 2023;120(13):e2216796120. https://doi.org/10.1073/pnas.2216796120.
Yuan Y, Wang L, Ge D, Tan L, Cao B, Fan H, Xue L. Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8(+) T cells. Cancer Lett. 2021;500:98–106. https://doi.org/10.1016/j.canlet.2020.12.012.
Ali A, Kim MJ, Kim MY, Lee HJ, Roh GS, Kim HJ, Cho GJ, Choi WS. Quercetin induces cell death in cervical cancer by reducing O-GlcNAcylation of adenosine monophosphate-activated protein kinase. Anat cell Biology. 2018;51(4):274–83. https://doi.org/10.5115/acb.2018.51.4.274.
Zhang C, Niu Y, Wang Z, Xu X, Li Y, Ma L, Wang J, Yu Y. Corosolic acid inhibits cancer progression by decreasing the level of CDK19-mediated O-GlcNAcylation in liver cancer cells. Cell Death Dis. 2021;12(10):889. https://doi.org/10.1038/s41419-021-04164-y.
Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits OGT activity in cells. ACS Chem Biol. 2015;10(6):1392–7. https://doi.org/10.1021/acschembio.5b00004.
Loi EM, Weiss M, Pajk S, Gobec M, Tomašič T, Pieters RJ, Anderluh M. Intracellular hydrolysis of small-molecule O-Linked N-Acetylglucosamine transferase inhibitors differs among cells and is not required for its inhibition. Molecules. 2020;25(15). https://doi.org/10.3390/molecules25153381.
Li M, Duan F, Pan Z, Liu X, Lu W, Liang C, Fang Z, Peng P, Jia D. Astragalus Polysaccharide promotes Doxorubicin-Induced apoptosis by reducing O-GlcNAcylation in Hepatocellular Carcinoma. Cells. 2023;12(6). https://doi.org/10.3390/cells12060866.
Yang R, Wang L, Wu Z, Yin Y, Jiang SW. How nanotechniques could vitalize the O-GlcNAcylation-Targeting Approach for Cancer Therapy. Int J Nanomed. 2022;17:1829–41. https://doi.org/10.2147/ijn.S360488.
Acknowledgements
We sincerely thank all the members who contributed to the study.
Funding
This work was supported by funding from the National Natural Science Foundation of China (No. 82203576), capital health research and development of special (2022-2-7083) and Beijing Municipal Natural Science Foundation (No. 7222100).
Author information
Authors and Affiliations
Contributions
Writing—original draft preparation, X.L.; writing—review and editing, J.W., Y.X., K.W., Y.T., and D.Y.; visualization, X.L.; supervision, Y.T. and D.Y.; project administration, J.W., Y.X., and K.W.; funding acquisition, Y.T. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, X., Wang, J., Xiang, Y. et al. The roles of OGT and its mechanisms in cancer. Cell Biosci 14, 121 (2024). https://doi.org/10.1186/s13578-024-01301-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-024-01301-w