
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease. The development of PD is closely linked to genetic and environmental factors, with GBA1 variants being the most common genetic risk. Mutations in the GBA1 gene lead to reduced activity of the coded enzyme, glucocerebrosidase, which mediates the development of PD by affecting lipid metabolism (especially sphingolipids), lysosomal autophagy, endoplasmic reticulum, as well as mitochondrial and other cellular functions. Clinically, PD with GBA1 mutations (GBA1-PD) is characterized by particular features regarding the progression of symptom severity. On the therapeutic side, the discovery of the relationship between GBA1 variants and PD offers an opportunity for targeted therapeutic interventions. In this review, we explore the genotypic and phenotypic correlations, etiologic mechanisms, biomarkers, and therapeutic approaches of GBA1-PD and summarize the current state of research and its challenges.
Similar content being viewed by others
Introduction
Parkinson’s disease (PD) is one of the most common neurological disorders characterized by motor symptoms such as resting tremor, bradykinesia, rigidity and postural instability, often accompanied by a variety of non-motor symptoms. The pathology of PD is characterized by the loss of dopaminergic neurons and the formation of α-synuclein (α-syn)-containing proteinaceous aggregates, known as Lewy bodies (LBs) and Lewy neurites [1]. The etiology of PD is not fully understood and its incidence increases with age, with genetic and environmental factors playing important roles in its onset and progression. With the rapid development of genetic testing technology, important advances have been made in the genetic aspects of PD [2]. To date, mutations in more than 20 genes and more than 90 common genetic risk loci have been reported to be associated with PD [2,3,4,5,6,7]. The roles of these genes and variants in PD remain controversial and require further validation and functional studies. Understanding the biological changes induced by these genetic variants is both important and challenging. Despite the complexity, studies of PD-associated genes have focused on a number of common pathogenic pathways, including lysosomal dysfunction, impaired intracellular trafficking, inflammatory signaling, and mitochondrial dysfunction. These pathways have been linked to dopaminergic neuronal damage [8].
Previous research has established a clear correlation between genes regulating lysosomal function and PD [9]. Recent studies revealed that over 50% of individuals with PD possess one or more putative damaging variants among the lysosome storage disorder (LSD) genes [9, 10]. Among the various genetic factors identified, the glucocerebrosidase gene (GBA1) is emerging as the predominant hereditary contributor to PD. GBA1 encodes the lysosomal enzyme glucocerebrosidase (GCase), which is responsible for breaking down glucosylceramide (GlcCer) [11]. In this review, we will summarize the genotypic and phenotypic correlations, etiological mechanisms, biomarkers, and therapeutic approaches for GBA1-PD as well as current challenges in its studies. Advances in GBA1-PD neuroimaging will not be included here, as they have been extensively reviewed elsewhere [12].
GBA1 and Gaucher’s disease (GD)
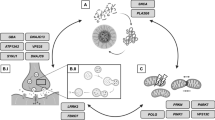
GD, initially reported by Philip Gaucher in 1882, is a prevalent LSD. The global prevalence of this autosomal recessive genetic disorder is 1.75/100,000. It is rare in the general population and has an impact on 1/40000–1/50000 births; however, it is relatively common among individuals of Ashkenazi Jewish descent, with approximately 1 in every 800 births being afflicted [13]. The pathogenesis of GD is attributed to biallelic variants of GBA1. Situated on chromosome 1q21, the GBA1 gene consists of 11 exons and 10 introns, spanning a length of 7.6 kb, and encodes the protein GCase of approximately 62 kDa, with three structural domains (Fig. 1). The functions and lysosomal targeting of GCase are supported by endogenous transporters and co-factors [14]. Lysosomal integral membrane protein 2 (LIMP2) is a type III transmembrane protein encoded by the SCARB2 gene. LIMP2 belongs to the CD36 family, is mainly distributed in the limiting membrane of lysosomes and is a receptor involved in the lysosomal translocation of GCase. LIMP2 binds GCase in the endoplasmic reticulum (ER) and reaches the lysosome via the Golgi system, where GCase dissociates from LIMP2 upon reaching the lysosome due to the acidic pH [15,16,17,18,19], where it subsequently interacts with the co-factor saposin C and negatively charged lipids for optimal catalysis activity (Fig. 2) [20]. Mutations in the GBA1 gene can lead to structural modifications of the GCase protein, resulting in decreased activity, stability, and protein levels of GCase, causing intracellular accumulation of its main substrate, GlcCer, in the reticuloendothelial system, which in turn gives rise to a spectrum of clinical symptoms, such as hepatosplenomegaly, anemia, thrombocytopenia, bone abnormalities, and neurodegeneration [21].

Schematic of the GBA1 gene, the encoded glucocerebrosidase (GCase) protein, and 3D structure of the protein. The most frequent GBA1 mutations, L444P, N370S, and E326K, are displayed. The 3D structure of the protein was generated using the PyMOL molecular graphics system

Glucosylceramide (GlcCer) synthesis and degradation pathway. Ceramide (Cer) is synthesized in the endoplasmic reticulum (ER) and then transferred to the cis-Golgi through vesicular transport. Cer is catalyzed by GlcCer synthase (GCS) to produce GlcCer, which is subsequently transported to the trans-Golgi side and transformed to lactosylceramide (LacCer), before being synthesized into complex glycosphingolipids (GSLs). GSLs are delivered to the cell membrane via vesicular transport. GlcCer breakdown is triggered by cell membrane endocytosis. The endosome transports GSLs to the lysosome during endocytosis. Saposin C (Sap-C) activates lysosome glucocerebrosidase (GCase) to degrade GlcCer to Cer and Glucose (Glc)
Globally, approximately 70%–98% of GD is caused by five relatively common mutations in GBA1, N370S, L444P, 84GG, IVS2 + 1 and RecNcil [22]. In GD, GCase activity is typically 10%–20% of that in normal individuals [23]. Different mechanisms can lead to reduced GCase activity, such as a loss of transcription/translation, misfolded GCase proteins that promote ER stress and activation of the unfolded protein response (UPR), inability of GCase to leave the Golgi apparatus, or loss of key amino acids in its catalytic domains. Similarly, different GBA1 variants may affect GCase activity through different mechanisms. For example, the L444P and N370S variants are associated with UPR activation and ER stress and may disrupt binding between GCase and saposin C, preventing saposin C activation of GCase [24,25,26,27]. The 84GG insertion is a frameshift mutation that typically results in premature stop codons and severely truncated or abnormally long proteins, leading to very low or even completely eliminated GCase activity [23].
GD exhibits a wide range of phenotypes that can be viewed as a spectrum, ranging from asymptomatic forms to severe organ damage and severe neurologic complications. Depending on the neurologic impairment, GD can be categorized into three subgroups. Type 1 is the predominant, non-neuronopathic form that may occur at any age. Types 2 and 3 generally have a more pronounced severity, often accompanied by different levels of neurologic damage [13]. These two subtypes are often referred to as neuronopathic GD. The acute neuronopathic type 2 GD is the rarest and most severe form. Patients with this type of GD usually die before the age of 2 years. The chronic neuronopathic type 3 GD is an intermediate form as it impairs functions of the central nervous system more slowly and gradually than type 2. Nonetheless, the entire spectrum of GD encompasses a diverse array of clinical manifestations, and neurologic impairment can manifest throughout the course of the disease [28].
GBAP1
Pseudogenes are DNA sequences with a high degree of homology to functional genes. Even though pseudogenes are considered ancient genes that have lost their function, they are still involved in the regulation of the expression of their parental genes at the transcriptional and translational levels [29]. The pseudogene of GBA1, GBAP1, is located approximately 16 kb downstream of this gene with a length of 5.7 kb [29, 30]. The coding region of the GBA1 gene shares 96% homology with the exonic region of GBAP1, while the region between the intron 8 and 3′ untranslated region has as high as 98% homology. The main exonic difference is a 55-bp deletion in exon 9, and the sequence similarity makes GBA1-specific sequencing difficult [31, 32]. The high degree of homology and proximity between GBA1 and GBAP1 facilitates recombination events caused by gene conversion, fusion, or duplication, and can give rise to a variety of structural variants [33, 34]. The recombination events between GBA1 and GBAP1 result in several different Gaucher mutations and mutant groups. The highest homology between GBA1 and GBAP1 is between exons 8 and 11; thus, the majority of disease-causing mutations are clustered at this position [35, 36]. Pseudogenes can act as competitive endogenous RNAs (ceRNAs) for their parental genes and compete for regulatory microRNAs (miRNAs). GBAP1 may act as a ceRNA to regulate GBA1 expression through miRNAs [37, 38]. The complexity of GBA1 gene rearrangements cannot be adequately captured using most current next-generation sequencing (NGS) methods, and Sanger sequencing should be used to achieve most accurate genotyping. Long-read NGS has improved ability to distinguish functional genes such as GBA1 from their pseudogenes; however, currently, its ability to identify recombinant alleles is still limited [39]. It is believed that with the rapid development of sequencing technology, the detection of GBA1 variants will be improved continuously [39, 40].
GBA1 associated with PD
Since the 1980s, clinicians have observed that many individuals diagnosed with type 1 GD exhibit concurrent PD, prompting investigations into the potential role of the GBA1 gene as a hereditary contributor to PD [41]. Over time, researchers have found that both biallelic and monoallelic mutations in GBA1 can increase the susceptibility to PD [42,43,44]. In 2009, a comprehensive meta-analysis involving 16 centers worldwide revealed an odds ratio of 5.43 for any GBA1 mutation between PD patients and the control group, underscoring GBA1 variants as a common risk factor for PD [45]. GBA1 mutation carriers face a significantly heightened rate of developing PD, ranging from 5 to 30 times [45,46,47,48], and this risk is influenced by factors such as age, ethnicity, and mutation type, with carriers of severe GBA1 mutations having a 3- to 4-fold higher risk than carriers of mild GBA1 mutations [48, 49]. Notably, there are disparities in the distribution frequency of GBA1 alleles among different ethnicities, with GBA1 variants accounting for 20%–30% of Ashkenazi Jews and 5%–15% of European patients [50]. In Chinese populations, GBA1 variants are found in 5.4%–8.4% of individuals with PD, compared to less than 1% in the general population [51,52,53,54].
Classification of GBA1 variants
Currently, over 300 GBA1 mutations and gene rearrangements have been reported. The classification of GBA1 variants is mainly based on the GD phenotype caused by the mutation. Mild mutations are typically associated with type 1 GD, whereas severe mutations lead to types 2 and 3 GD. Certain variants of the GBA1 gene do not result in GD when present in homozygous carriers; however, these variants elevate the susceptibility to PD and are therefore categorized as “risk” variants. Additionally, a growing number of “unknown” variants have been reported, owing to the limited knowledge of their pathogenicity concerning GD or PD. Overall, 5.9% are classified as mild (e.g., N370S, R496H), 22.6% as severe (e.g., L444P, 84GG, IVS2 + 1, V394L, and D409H), and 0.8% as risk (E326K, T369M, and E388K), with the remaining categorized as unknown (e.g., A456P, K(-27)R, R39C, and R44C) [49]. It is important to note that the relationships between genotype and phenotype are not universally applicable, given that there are some outliers. Parlar et al.[49] recently compiled an inventory of all GBA1 mutations found in patients with PD and developed an online viewer (https://pdgenetics.shinyapps.io/gba1browser/) to search for these variants. The browser summarizes all reported GBA1 variants in PD to date, and in addition to classifying the variants as described above, they also categorize the pathogenicity of GBA1 variants according to the American College of Medical Genetics and Genomics (ACMG) classification of variant in GD (Additional file 1: Table S1). Notably, the distribution of GBA1 variants varies significantly in different ethnic groups. The most common GBA1 mutation in PD among Ashkenazi Jewish is the N370S mutation. East Asians and South Asians predominantly exhibit the L444P mutation, while Europeans have a higher prevalence of the E326K and T369M GBA1 mutations [51, 55,56,57].
Penetrance of GBA1 mutations
Penetrance is the ratio of all individuals carrying a disease-associated allele to those affected by the disease. Large-scale sequencing and genotyping studies provide a powerful means to understand the penetrance of pathological mutations/genotypes. The main methods used to estimate penetrance of mutations in PD are the Kaplan–Meier method for unrelated individuals and the kin-cohort method within families [58]. The penetrance of GBA1 increases with age, with the reported penetrance for GBA1 heterozygous mutations ranging from 10% to 30% at 80 years of age, suggesting that the majority of GBA1 heterozygous mutation carriers will never develop PD [59,60,61]. Similarly, follow-up of nine GBA1 L444P/R non-manifesting carriers for up to 10 years did not find any of them developing PD [62]. Notably, a study in patients with Ashkenazi Jewish heritage showed an increased risk for PD in both patients with GD (carrying 2 mutant GBA1 alleles) and heterozygote carriers (carrying 1 mutant GBA1 allele); however, the overall risk for developing the disease was similar between the two groups of patients, suggesting that the number of mutant alleles may not affect the penetrance [43].
The reason for the relatively low rate of PD phenotypic transformation in individuals carrying GBA1 mutations remains elusive. Genetic factors may be involved. The GBA1-associated risk is significantly influenced by the genetic risk score. Specific locus variants, such as SNCA and CTSB, could also influence the risk of PD by interacting with GBA1. In addition, environmental, aging, and epigenetic factors are also involved [38, 63, 64]. Therefore, in studies targeting GBA1 penetrance, a comprehensive assessment of genetic factors should be performed [63, 65], taking into account the influence of non-genetic factors such as environmental factors, aging, demographics/ethnicity and diet. Large prospective cohort studies and meta-analyses in GBA1 non-manifesting carriers would allow for more accurate estimates of GBA1 penetrance [43].
Genotype–phenotype characteristics of GBA1-PD
The relationship between genotype and phenotype is complex. Although the general phenotype of GBA1-PD resembles idiopathic PD, patients with GBA1-PD exhibit distinct clinical features as a group [66]. GBA1 mutations accelerate the neurodegenerative process, resulting in significantly impaired dopaminergic functions and a more severe clinical phenotype [67, 68]. Specifically, GBA1-PD tends to have an earlier onset [55, 69], is more likely to have a family history, and is associated with more severe motor and non-motor symptoms. In addition, patients with GBA1-PD have more rapid disease progression and higher mortality (Fig. 3) [70, 71].

Stage characteristics of GBA1-PD. PIGD, postural instability and gait difficulties
A dose-dependent relationship between GBA1 variants and the age of PD onset may exist. Compared to heterozygous and mild or risk variant carriers, carriers of biallelic GBA1 mutations (either homozygous or compound heterozygous) or severe variants tend to have an earlier age of onset [43, 48, 72, 73]. Prodromal symptoms of PD may be more prevalent among GBA1 variant carriers [28, 74]. GBA1 non-manifesting carriers may present with typical prodromal symptoms of PD, such as rapid eye movement sleep behavior disorder (RBD), hyposmia, cognitive decline, and fine motor symptoms, and these symptoms may progressively worsen over time [28, 75,76,77,78,79,80].
Individuals harboring GBA1 mutations exhibit an increased incidence of motor complaints and a higher likelihood of postural instability and gait difficulties, along with gait and balance disorders [81]. They often experience more rapid progression of motor symptoms [82]. Additionally, patients with GBA1-PD experience motor fluctuations and dyskinesias earlier [47, 73, 83]. In terms of non-motor symptoms, patients with GBA1-PD are more susceptible to developing RBD and autonomic complaints such as constipation, and a greater degree of olfactory loss [55, 68, 73, 79, 84, 85]. They are six times more likely to develop dementia than non-carriers, and have more rapid progression of cognitive impairment [68, 71, 73, 86,87,88], particularly in visuospatial ability, executive function, and working memory [89, 90]. The more extensive neocortical Lewy body-type lesions in patients with GBA1-PD, and the higher rate of GBA1 mutation in patients with dementia with Lewy bodies (DLB), also reflect, to some extent, the close relationship between PD and dementia [91, 92]. Additionally, there is an increased prevalence of psychological symptoms, including hallucinations, delusions, and impulsive behavior in patients with GBA1-PD [84]. In terms of outcomes, GBA1 mutation carriers have twice the risk of death than non-carriers, die 1 year earlier, and may have lower survival rate when carrying severe mutations [71].
Modification of GBA1-PD by other genes
Patients carrying both LRRK2 and GBA1 gene variants have a milder phenotype and exhibit fewer motor, non-motor, and dementia symptoms compared to patients carrying a single variant allele of GBA1, suggesting that LRRK2 gene mutations may have a beneficial effect in individuals with GBA1 gene mutations [93, 94]. However, the exact underlying mechanism remains incompletely understood. One possible explanation is the presence of crosstalk between the two proteins. In dopaminergic neurons derived from PD patients with LRRK2 mutations, GCase activity is diminished, and inhibition of the kinase activity of LRRK2 leads to increased GCase activity, with the LRRK2 substrate Rab10 as a key mediator in the regulation of this process [95]. Furthermore, inhibition of LRRK2 kinase reverses some lysosomal impairments and inflammatory responses observed in astrocytes with GBA1 mutations [96].
Genes closely associated with Alzheimer’s disease (AD) can also influence the phenotype of GBA1 carriers. For example, individuals carrying mutations in both GBA1 and the APOE epsilon 4 allele show a higher risk for cognitive impairment [90]. This combination results in a five-fold elevation in the likelihood of developing PD dementia and accelerates the rate of cognitive deterioration [97]. Bridging Integrator 1 (BIN1), a second major risk locus for late-onset AD, plays a crucial role in mediating tau pathology, endocytosis, inflammation, calcium homeostasis, and apoptosis [98,99,100]. Alterations in the BIN1 locus may also influence the age of onset of GBA1-PD [101].
It is important to recognize that the genotype–phenotype relationships are complex and highly heterogeneous. To fully understand the impact of GBA1 variants on the natural history of PD, it is necessary to examine the prodromal symptoms of PD in GBA1 non-manifesting carriers. Long-term prospective cohort studies of such risk groups can help identify clues to the transformation of PD. Evaluating the impact of GBA1 variants on various types of motor and non-motor symptoms, motor complications, death, and other outcomes of PD is significant to elucidate the trajectory of GBA1-PD. Considering the impact of different mutation types on the GBA1 phenotype, it is necessary to stratify patients according to the GBA1 mutation type, include more homozygous and complex congenic carriers (GD-PD), and exclude common PD-associated genes such as LRRK2 and PRKN from clinical trials [71, 73, 82, 102, 103]. Additionally, it is necessary to consider the impact of medication factors and the on/off status of clinical symptoms when evaluating patients. In addition to clinical scale assessments, the use of highly specific instruments or objective measures with cutting-edge technologies such as biochemical, neuroimaging, and artificial intelligence approaches will improve the sensitivity to subtle changes [61, 104, 105]. It is worth noting that the disease phenotype is influenced by a variety of factors, such as the environment, different protein expression thresholds, or other modifier genes. Only by comprehensively considering these important phenotypic regulators can the complex relationship between genotype and phenotype be finally unraveled [25, 106].
Mechanism of GBA1 involvement in PD
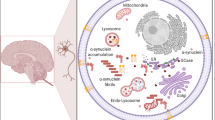
In recent years, research has delved into the role of mutant GBA1 in the pathogenesis of PD. GBA1-PD does not strictly adhere to Mendelian laws, as both gain-of-function and loss-of-function mutations have been associated with an elevated risk of developing PD. The mechanisms underlying the increased risk of PD from GBA1 variants remain incompletely understood. Research progress has indicated the involvement of dopaminergic neuronal damage, α-syn aggregation, lipid metabolism disorders, autophagy-lysosome dysfunction, ER stress, mitochondrial damage, and inflammatory responses (Fig. 4). In the following, we will discuss each of the listed mechanisms separately in detail.

Mechanism of mutant glucocerebrosidase (GCase) in PD. a Under healthy conditions, mRNA from the GBA1 gene travels from the nucleus to the endoplasmic reticulum (ER). GCase is produced in the ER, transferred to the Golgi apparatus by binding to the protein LIMP2, and then fused with lysosomes, where GCase is activated to exert its hydrolytic effect. Usually, α-syn can be phagocytosed by autophagosomes and then targeted to lysosomes, where GCase interacts with α-syn to promote its degradation. b GBA1 mutations lead to impaired GCase activity, and GCase proteins misfold and lodge in the ER, initiating ER stress. Lack of GCase in lysosomes impairs the autophagic lysosomal pathway (ALP), causing accumulation of lipid substrates such as GlcCer in lysosomes and α-syn aggregation. This accumulation prevents GCase transfer from the ER/Golgi to the lysosome, exacerbating lysosomal dysfunction. GCase deficiency and reduced ALP function may cause mitochondrial dysfunction, resulting in reactive oxygen species (ROS), decreased ATP generation, and aberrant mitochondrial morphology. In addition, GCase deficiency, accumulation of lipids, and α-syn may activate microglia and cause neuroinflammation. All of these contribute to cell death and the development of PD
GCase and dopaminergic neurons
A central question regarding GBA1-related PD is how the loss of GCase activity predisposes dopaminergic neurons to the accumulation of α-syn [107]. Although there may be a bidirectional feedback loop between GCase deficiency and α-syn accumulation, it does not explain the selective vulnerability of midbrain dopaminergic neurons [108]. However, there is much evidence to support a strong link between GBA1 variants and dopaminergic neurons. For example, the GCase enzyme activity was found to be reduced in dopaminergic neurons from patients with GBA1-PD, and there is a significant loss of dopaminergic neurons in the brain [107, 109, 110]. In Drosophila, mutant GCase and α-syn have synergistic deleterious effects on dopaminergic cells [111]. Studies using many cellular/neuronal models have also shown that GBA1 mutations can exacerbate the bioenergetic burden on dopaminergic neurons via disruption of lipid homeostasis, release of inflammatory factors, oxidative stress, mitochondrial dysfunction, and impaired calcium homeostasis [107, 108, 112, 113]. However, downregulation of GCase expression alone is not sufficient to cause the key pathological features of PD in vivo. Synergization with α-syn overexpression is required for GCase deficiency to drive nigrostriatal neurodegeneration [110]. A study by Henderson et al. [114] showed that reduced GCase activity alone does not lead to the aggregation of α-syn, but rather functions by enhancing the aggregation of pre-existing misfolded α-syn, and this modulatory effect is not dependent on the neuronal type. Indeed, α-syn and GCase are widely expressed in the brain, affecting different cell types [108, 115]. Notably, Burbulla et al. [116] showed that although increased α-syn affects GCase transport and lysosomal function in both dopaminergic and non-dopaminergic cells, oxidized dopamine accumulation was only observed in dopaminergic cells, which could reduce wild-type or mutant GCase activity [116,117,118]. In GBA1-PD organoid studies, NURR1, a transcription factor essential for the differentiation, maturation, and maintenance of midbrain dopaminergic neurons, was found to be significantly reduced, and this reduction led to reduced dopamine production and increased susceptibility of these neurons [119]. It has also been hypothesized that mutated GCase proteins do not fold sufficiently in the ER, leading to protein accumulation in the cellular compartment, which induces dopaminergic neuronal response to stress, causing their damage and death [120].
The vicious cycle between GCase and α-syn
PD-related mutations in GBA1 may impair GCase activity. GCase deficiency increases the aggregation and spread of α-syn and promotes neurodegeneration in PD. Numerous basic studies have demonstrated the close relationship between GCase and α-syn (Table 1) [114, 121, 122]. Examination of mutations across several vertebrate species indicates a potential co-evolutionary relationship between GCase and α-syn, as alterations in either protein can hinder their interaction. Decreased GCase activity may lead to oligomerization of α-syn aggregates, and accumulated α-syn may also impede GCase migration from the ER to the lysosome, resulting in a decrease in GCase activity, leading to a pathogenic feedback loop [114, 121, 123].
Additionally, α-syn can directly interact with GCase under acidic conditions, further inhibiting GCase function [124]. Reduced GCase activity can potentially enhance α-syn phosphorylation at serine 129 and oligomerization in the brains of aged monkeys [125]. Various risk variants of GBA1 may result in reduced GCase activity through several mechanisms, including direct reduction of enzyme activity, failure to adhere to the ER quality control systems resulting in proteasome degradation, retention within the ER or Golgi apparatus, and incorrect ligation of LIMP2 or saposin C [25, 126]. Studies conducted on human samples have revealed a negative correlation between pathological α-syn and GCase activity in brain tissues of PD patients and a negative correlation between GCase activity in leucocytes and plasma oligomeric α-syn in patients with GD [123, 127, 128]. DLB, characterized by the accumulation of aggregated α-syn protein in LBs and Lewy neurites as the main pathologic features, is also strongly associated with GCase, which has been detected in LBs [129, 130]. Decreased GCase activity and elevated glycosphingolipid (GSL) level may exacerbate Lewy pathology [131]. These findings collectively support the close relationship between α-syn and GCase. However, it is important to note that the incidence of PD does not appear to increase with decreased enzyme activity, and a considerable proportion of patients with GD never progress to PD. Therefore, considering that the loss of enzyme activity is not the only cause of PD, further investigation is needed to explore other mechanisms by which GBA1 mutations contribute to α-syn accumulation.
Lipid metabolism disorder
GSLs are found in cell membranes and play a critical role in maintaining membrane function and facilitating cellular communication in a variety of tissues, including the brain. However, they are not restricted to the intracellular compartment, but also act in the extracellular space (e.g., plasma) through unknown mechanisms. GlcCer and glucosylsphingosine (GlcSph), substrates of GCase, both belong to the class of GSLs. GlcCer is abundant in the brain and plays an essential role in intracellular membrane trafficking, signal transduction, and cell proliferation [132, 133]. Mutations in the GBA1 gene affect the metabolism of GSLs, and dysfunction of the enzyme GCase results in the intracellular accumulation of its upstream substrate GlcCer, which can leave the lysosome to enter the cytoplasm or remain in the lysosome and be converted to GlcSph and released into the cytoplasm [134, 135]. Sphingolipids, including GlcCer and GlcSph, have been reported to affect the secondary structure and aggregation kinetics of α-syn under acidic or neutral pH conditions in vitro and to exacerbate Lewy pathology by affecting the fluidity of lysosomal membranes and promoting the formation and aggregation of α-syn fibers [121, 131, 136,137,138,139,140,141,142,143]. Conversely, oligomers of α-syn impede GCase migration from the ER to the Golgi, reduce GCase activity, and affect substrate accessibility and turnover, leading to further accumulation of GlcCer [121, 144, 145].
Animal studies have shown that reducing GSLs with GlcCer synthase inhibitor slows the progression of synucleinopathy [146]. Despite increasing data on the effects of GBA1 on lipid metabolism and PD pathogenesis, there is still a long way to go to fully understand the complex interactions between genetic background, disease status, GCase activity, lipid metabolism, and protein pathology.
Impaired autophagy-lysosome pathway (ALP)
Lysosomes are acidic organelles that degrade toxic cellular contents through enzymatic degradation and autophagic pathways. Many neurodegenerative disorders, including PD, are associated with lysosomal abnormalities. Autophagy is the cellular process through which an organism removes proteins and degrades dysfunctional organelles within the cell, which may prevent cytotoxicity and cell death. Since α-syn undergoes degradation within lysosomes, dysfunction in the ALP could hinder the clearance of α-syn, leading to its aggregation into harmful oligomers. Consequently, this can exacerbate the cycle of impaired autophagy and lysosomal function [11, 147]. Evidence from experimental PD models suggests a strong association between GBA1 and ALP regulation [95, 148].
The autophagy pathway can be categorized into macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy is the primary regulated type of autophagy that responds to environmental and physiological cues, microautophagy involves the direct engulfment of cytoplasmic contents by lysosomes through membranous invaginations, and CMA is a protein-exclusive type of autophagy involving chaperone-assisted translocation of substrate proteins across the lysosomal membrane [149, 150]. Macroautophagy and CMA are particularly relevant to PD [151]. Cells and animal models deficient in GCase show impairment of ALP, with decreased lysosomal GCase activity for macroautophagy and CMA and interfering with α-syn clearance [152]. Mice deficient in the lysosomal enzyme β-hexosaminidase exhibit α-syn accumulation in neurons [153]. Studies of fibroblasts from PD patients carrying the N370S GBA1 variant revealed increased autophagic structures in the variant cells and accumulation of SQSTM1/p62 [154]. PD patients exhibit CMA defects in their brains, with autopsies indicating a significant decrease of CMA markers in specific regions and accumulation of autophagy-associated proteins in the LBs [155]. Single-cell transcriptional analyses and proteomics have confirmed decreased CMA activity and proteomic alterations in GBA1-PD [156]. Modulating autophagy can ameliorate disease pathology. For example, inducing autophagy using the autophagy activator rapamycin or promoting GCase translocation into lysosomes with the GCase chaperone isofagomine can reverse α-syn accumulation [157]. Overexpression of Beclin 1, which stimulates autophagy, can ameliorate the neurodegenerative pathology in PD animal models [158].
In addition to macroautophagy and CMA damage, GCase depletion has been found to cause lysosomal dysfunction in mouse neurons and Drosophila brains [121, 159]. GBA1 D409V-knock-in mouse astrocytes exhibit extensive defects in lysosomal morphology and function, including reduced lysosome numbers, fewer acidic vesicles, and reduced lysosomal degradative activity [146, 148]. GCase-deficient Drosophila exhibit many abnormally expanded lysosomes in the brain [160]. The mechanisms by which the loss of GCase activity affects lysosomes are unclear. GCase dysfunction impairs the lysosomal compartment, establishing a lysosome–plasma membrane axis that leads to structural changes of the plasma membrane and alterations of intracellular signaling pathways. At the same time, accumulation of GlcCer or GlcSph, substrates of GCase, could contribute to lysosomal dysfunction and toxicity [161, 162].
Studies in PD patients have revealed extensive lysosomal dysfunction. Variants in LSD genes have been detected in both PD and GBA1-PD patients [163], and LSD patients (GM1 and GM2 gangliosidosis, neuronal ceroid lipofuscinosis, and Fabry disease) can exhibit typical PD symptoms [164]. To date, many genetic loci related to autosomal/lysosomal function have been identified as risk factors for familial and sporadic PD. Large-scale meta-analyses of GWASs have implicated several dozens of loci associated with lysosomal functions and pathways in PD pathogenesis [5, 6, 165,166,167]. Multiple lines of evidence suggest that genetic variants of lysosomal genes contribute to PD more broadly. In addition to GBA1, other genes involved in LSD (e.g., SMPD1, SCARB2, and GALC) can also increase the risk of PD. Other lysosomal genes (ATP13A2, LAMP1, TMEM175, and VPS13C) that increase the burden of variants in PD patients have recently been reported [163, 168,169,170,171]. Growing evidence also highlights the impact of multifunctional lysosomal proteins (e.g., LIMP2, prosaposin, progranulin, and cathepsin D) on PD [172]. LIMP2 deficiency reduces GCase activity, impairs autophagy/lysosomal function, and leads to lipid storage and α-syn accumulation, resulting in neurodegeneration, inflammation, and apoptosis. Conversely, overexpression of LIMP2 reduces α-syn levels [173]. Changes in lysosomal enzymes associated with various degradation pathways have been observed in body fluids and tissues, including the brain, the cerebrospinal fluid, and fibroblasts of PD patients [131, 174,175,176,177,178]. For example, expression of the major lysosomal protease cathepsin D is reduced in nigral neurons in PD patients, especially in neurons containing α-syn inclusions [179]. At the same time, altered activities of cathepsin D, cathepsin E, α-fucosidase, β-hexosaminidase, and β-galactosidase are observed in the cerebrospinal fluid of patients with PD [175,176,177].
ER stress
ER stress may cause several neurodegenerative disorders. GBA1 abnormalities can also induce PD through ER dysfunction. ER is essential for protein folding, lipid synthesis, and calcium storage. Dysregulation of ER homeostasis can activate the UPR, initiating a cascade of significant stressors within the ER. This response aims to restore the equilibrium but may activate pathways that lead to cellular demise [180].
GCase is synthesized on polyribosomes bound to the ER and subsequently translocated into the ER. Within the ER, the protein undergoes N-linked glycosylation at four asparagine residues and quality control [120]. Mutant GCase proteins are misfolded, resulting in varying degrees of ER retention, triggering ER stress, UPR, and Golgi fragmentation [181]. UPR and ER stress can result in autophagy damage, increased apoptosis, and lysosomal insufficiency, weakening the protective role of autophagy against synucleopathies and ultimately contributing to PD development [154]. ER stress and UPR activation markers are elevated in GBA1-mutant brain tissues as well as cellular and animal models [182,183,184,185,186], supporting the conclusion that GBA1 mutations cause abnormal changes in ER function. In α-syn-overexpressing neurons, the inability of LIMP2 to bind to GCase leads to increased GCase retention in the ER and decreased GCase activity in the lysosome [121, 187].
Mitochondrial dysfunction
Mitochondrial turnover is essential for neuronal protection. Failure to remove damaged mitochondria can lead to the accumulation of reactive oxygen species (ROS) and free radicals, causing neural damage. The onset of PD is associated with mitochondrial dysfunction, and PD-associated genes are linked to mitochondrial morphology and function. Increasing evidence shows that decreased GCase activity may disrupt mitochondrial function [188,189,190].
Impaired mitochondrial function and morphology, reduced ATP production, and increased oxidative stress have been observed in GCase-deficient cells, animal models, and PD patients [189, 191, 192]. In SH-SY5Y neuroblastoma cells, inhibition of GCase activity by conduritol B epoxide (CBE) significantly reduced mitochondrial membrane potential (Ψm). Ψm, generated by the mitochondrial electron transport chain, plays a significant role in ADP phosphorylation into ATP [193]. GD fibroblasts show mitochondrial abnormalities associated with impaired macroautophagy flow [194]. Mitophagy activation decreases the colocalization of mitochondria and LC3 in GBA1-knockout cells [195]. Abnormal mitochondrial morphology and function, increased ROS, reduced ATP production, autophagosome accumulation, and impaired mitophagy have been observed in GBA1 L444P heterozygous mutant mouse models, GBA1 knockout mice, and GCase-deficient Drosophila brains, findings that are consistent with those observed in patients with GBA1-PD [186, 196,197,198]. Mitochondrial accumulation of α-syn was seen in cortical neurons in GD mice and resulted in abnormal mitochondrial morphology and function [199].
Inflammatory response
GBA1 mutations may be associated with inflammation [200,201,202]. GD is accompanied by significant alterations associated with inflammation, including elevated proinflammatory factors and chemokines, and activation of microglia, astrocytes, and T cells [203]. Reduction of GCase activity by CBE treatment in animals induced strong neuroinflammatory responses, complement activation, microglial activation, and insoluble α-syn aggregation [204,205,206]. Similarly, inflammatory mediators are increased in the plasma from GBA1-PD patients [207], and levels of interleukin (IL)-8, monocyte chemoattractant protein-1, and macrophage inflammatory protein-1 alpha are significantly associated with the GBA1 mutation genotype [208]. Macrophages from patients harboring the homozygous GBA1 N370S mutation exhibit increased secretion and inflammatory activation of IL-1β and IL-6 [209]. GCase activity inhibition, α-syn aggregation, and monocyte lineage-mediated inflammation lead to pathogenic cascade responses. These responses can induce neuronal damage and cell death and ultimately drive PD pathology [210].
Advances in cellular and animal models of GBA1-PD
Establishing suitable models to study the pathogenesis of PD remains a challenging task. Human induced pluripotent stem cell (iPSC) technology provides an opportunity to study the role of genetic mutations in the pathogenesis of PD. The iPSC technology enables studies of patient-derived cells in the genetic context of the patient’s disease [108]. Isogenic gene-corrected cell lines help uncover the genetic mutation-associated abnormalities that are involved in disease onset and neuronopathic phenotypes [211, 212]. Cell models generated from neural derivatives of iPSCs based on GBA1 mutations provide a unique tool for studying pathological changes within PD cells [213]. Another in vitro approach for studying human brain functions is the use of brain organoids. Compared to standard 2D cultures, iPSC-derived 3D organoids have greater similarity to the in vivo cellular tissues and organ structures, allowing for the assessment of specific mutations in more complex neural-specific models [25, 119, 214]. Patient-derived midbrain organoids carrying specific genetic mutations represent a more suitable tool to generalize the disease process occurring in PD patients [215, 216]. Midbrain organoids mimic early embryonic neurodevelopment and are ideal models for studying PD-related neurodevelopmental phenotypes. Midbrain organoids with GBA1 N370S mutation show severely impaired neurodevelopment [119]. In terms of animal models, GBA1-associated PD models have been developed in combination with overexpression of α-syn [217,218,219]. Progranulin (PGRN), encoded by GRN, is a novel GCase modifier. In vivo and in vitro data suggest that PGRN deficiency plays a crucial role in the initiation, progression, and regulation of GD associated with GBA1 mutations. Some researchers have crossed PGRN-deficient mice (Grn−/−) with GbaD409V/D409V (Gba9v/9v) mice to produce Grn−/−Gba9v/9v (termed PG9V) mice. This mouse model overcomes many of the limitations of existing models, as it exhibits the neurodegenerative manifestations of GD and PD, with an exacerbated GD phenotype [220].
Biomarker studies of GBA1-PD
Objective biomarkers for GBA1-PD are still lacking, but dysfunction in the GCase pathway offers insights into potential biomarkers for PD. GCase activity and sphingolipid content can be determined from specimens such as brain tissue and blood. These biomarkers can potentially improve patient management and counseling in clinical settings, and serve as metrics for evaluating the effectiveness of GCase-targeting therapy [24].
GCase enzymatic activity
Since 2012, studies using brain autopsy and dried blood spot testing have reported reduced GCase activity in PD patients [131, 176,177,178, 187, 221, 222]. Since most patients with GBA1-PD still have a wild-type GBA1 allele, GCase activity is only slightly reduced when compared with controls, and the extent of this reduction may be related to the variant classification [221,222,223]. As seen in brain tissue assays, there is a general decrease in GCase activity in patients with GBA1-PD, with the most significant decrease in the substantia nigra [187]. However, findings on GCase activity have been inconsistent and, rather surprisingly, reduced GCase protein level and enzyme activity have also been reported in patients with sporadic PD [123, 131, 174, 175, 187]. Compared to brain biopsy, blood samples can be collected prospectively and longitudinally, and the procedure is less invasive than lumbar puncture. Whole blood samples (GCase is active in leukocytes, not plasma) are important for detecting GCase, with numerous studies utilizing dried blood spots to test GCase activity [222]. In 2015, the first study on dried blood spot GCase activity in GBA1-PD was conducted [221], showing that the GBA1-PD heterozygotes exhibit lower GCase activity than controls. The enzymatic activity decrease shows a dose effect, with GCase activity decreasing with increased number of GBA1 variant alleles. However, the exact relationship of enzymatic activity with the severity of GBA1 mutations and the clinical phenotype, is debated. Similar to the results from brain tissue assays, GCase enzymatic activity in blood is reduced even in PD patients lacking GBA1 mutations [221, 222]. It is noteworthy that there is cellular heterogeneity in GCase activity, as a significant reduction in GCase activity has been observed in monocytes from PD patients, whereas the GCase activity in lymphocytes is not significantly reduced [224].
LRRK2 affects the GCase enzymatic activity
LRRK2 mutations are a common genetic etiology of PD. A study examined the GCase activity among carriers of LRRK2 variants and in GBA1-PD patients. Surprisingly, the LRRK2 G2019S carriers showed higher GCase activity than non-carriers [221]. This phenomenon was also observed in the peripheral blood mononuclear cells (PBMC) of individuals with LRRK2 mutations [225]. The mechanisms underlying the increased GCase activity in LRRK2 variant carriers remain unclear. One hypothesis suggests that LRRK2 mutations might result in the expansion of lysosomal compartments, a phenomenon associated with increased GCase activity [226]. Alternatively, LRRK2 mutation could impact reverse retromer function, potentially altering GCase localization and turnover [222, 227]. However, a recent study on GCase activity in brain tissues did not show any significant changes in the enzymatic activity in PD patients with LRRK2 mutations [131]. Conversely, another study found decreased GCase activity in the fibroblasts and dopaminergic neurons derived from iPSCs from LRRK2 mutation carriers, and this reduction could be reversed by inhibiting LRRK2 kinase [95]. Thus, the LRRK2 kinase activity affects GCase activity differently depending on the test medium, the cell type, and the technique employed.
Sphingolipid metabolism
Sphingolipid metabolism can be examined in plasma. However, findings from plasma assays are ambiguous and inconsistent [228, 229]. A pooled data analysis of five studies explored the variability of various GSLs across different samples (plasma, PBMC, and cerebrospinal fluid). Results revealed that elevated plasma levels of GlcCer can help differentiate among idiopathic PD, GBA1-PD, and healthy individuals, with the GlcCer C24:1 isoform being the most effective in differentiating GBA1-PD from idiopathic PD and healthy individuals [139]. A study of plasma GlcSph, GlcCer, ceramide, and four other lipids in carriers of the GBA1 N370S mutation found that GlcSph was the only variable associated with GBA1 mutation status [230]. Several brain tissue studies have found elevated GSLs in idiopathic PD. However, these alterations have been observed with inconsistent conclusions across specific brain regions and age groups. For instance, it has been reported that GlcCer and GlcSph accumulate in non-GBA1-PD [229, 231], whereas no accumulation occurs in putamen [162]. It has also been shown that GlcSph levels are elevated in GBA1-PD and idiopathic PD, while GlcCer levels are unchanged [131]. Similarly, an analysis of 251 lipids, including 95 sphingolipids, in brain tissues showed changes in ganglioside levels in non-dopaminergic areas of the GBA1-PD brain. In contrast, changes in other lipid classes, including GlcCer, were minimal [232]. Conversely, quantitative analysis of sphingolipids in cerebrospinal fluid samples revealed a considerable rise in GlcCer levels and a reduction in the sphingomyelin fraction, a crucial downstream metabolite [233].
In summary, available evidence does not support the use of reduced GCase activity, or GlcCer or GlcSph accumulation as a reliable biomarker of PD. While GBA1 variant carriers exhibit a trend toward decreased GCase activity in brain tissues, blood and fibroblasts, as well as decreased mean intracerebral GCase activity, there is a substantial overlap between patients and controls. GCase activity is highly variable in populations. Besides the type of GBA1 mutation, GCase activity is also related to factors such as the sample source and type, sample storage condition and duration, the number of times the samples are frozen and thawed, the assay method used, and other genetic or environmental factors [170, 222, 234,235,236,237]. Although the conclusions have not been entirely consistent, current evidence supports the notion that loss of GCase function may be one of the pathogenic mechanisms of PD and is a potential therapeutic target.
There are still many questions about GCase activity, such as the relationship among GCase activity, the disease state, the genetic status, the associated lipid metabolism, and protein pathology in various regions of the brain [131]; and the correlation among GCase activity in dried blood spots, cerebrospinal fluid, and the brain parenchyma. It is important to note that different cells could have different levels of GCase, and cell-based analysis might provide more accurate information for the detection of GCase activity [78, 224, 238]. Further cell-based and animal model studies will be beneficial for elucidating the GCase biology [118]. Likewise, contradictory findings regarding GlcCer and GlcSph concentrations dampen our enthusiasm on their potential as biomarkers for GBA1-PD. Some researchers have proposed that since the GCase reaction is part of a complicated sphingolipid cycling pathway, analyzing the consequences of reduced GCase activity on the overall sphingolipid cycling pathway and sphingolipid flux is essential for drawing meaningful conclusions [138]. Investigating cellular pathways beyond GlcCer or GlcSph is necessary to identify the molecular linkage between GBA1 mutations and PD [232].
Treatment
The association between GBA1 variants and PD presents promising opportunities for targeted PD therapies aimed at the GCase pathway. Patients with PD who carry GBA1 variants often exhibit a more aggressive disease progression phenotype, necessitating more intensive treatment strategies to counteract poor prognosis. The basic principle of clinical trials targeting GBA1-PD is to regulate GSL turnover and correct defects in cellular GCase. Specific approaches include enzyme replacement therapy (ERT), substrate reduction therapy (SRT), pharmacological small-molecule chaperones, and gene therapies [24, 126, 239].
ERT and SRT
ERT and SRT have proven effective therapeutic approaches for GD (Fig. 5). ERT involves administering recombinant GCase enzymes to replace dysfunctional GCase in patients. These enzymes typically possess modified terminal mannose residues, enhancing their targeting and macrophage absorption [180]. However, the recombinant GCase cannot penetrate the blood–brain barrier (BBB), resulting in limited efficacy in addressing neurologic symptoms associated with GD. In contrast, SRT prevents the overaccumulation of GlcCer by inhibiting GlcCer synthase, thereby reducing substrate accumulation [180]. Similar to ERT, SRT also faces challenges in BBB crossing.

Enzyme replacement therapy (ERT), substrate reduction therapy (SRT), and molecular chaperone therapy targets. Glucosylceramide synthase (GCS) synthesizes glucosylceramide (GlcCer) from ceramide (Cer), and glucocerebrosidase (GCase) degrades GlcCer to Cer. ERT increases GCase, and SRT decreases GlcCer in GBA1-PD. Molecular chaperones increase the stability of mutant GCase
Brain-penetrating GlcCer synthase inhibitors such as GZ667161 and venglustat can reduce substrates for GCase enzymes in the brain, decrease α-syn level in experimental animals, and improve animal behavior [240, 241]. Venglustat is safe and well tolerated in PD patients harboring GBA1 variants (ClinicalTrials.gov ID: NCT02906020, Moves-PD). Venglustat effectively eliminated GlcCer accumulation; however, it did not show a beneficial therapeutic effect [242, 243], suggesting that GlcCer accumulation is not a major pathogenetic mechanism and thus not a desirable pharmacological therapeutic target for PD, and that the complex role of GBA1 in PD extends beyond substrate accumulation [118, 242, 244, 245]. It is thus necessary to further decipher novel pathway interactions based on GCase–α-syn at the molecular, cellular, and systemic levels.
Pharmacological small-molecule chaperones
Various small molecules with GCase-activating properties have stabilizing or ameliorating effects on neurological symptoms [246]. More recently, several medications have emerged as potential enhancers of GCase activity [247,248,249,250,251]. Among these, chaperonins are of interest for their ability to enhance lysosomal GCase activity (Fig. 5), as they can penetrate the BBB and correct the folding of GCase in the ER, facilitating its translocation to the lysosome [15]. Preclinical studies have been conducted in GBA1-PD models [251,252,253]. Ambroxol has been shown to enhance GCase activity and is the most promising small-molecule chaperone targeting the GCase pathway [254, 255]. Ambroxol can cross the BBB, and increase GCase level and activity as well as GBA1 mRNA expression in fibroblasts of patients with PD. It can also increase the expression/activity of lysosomal cathepsins, LIMP2 and saposin C, and modulate multiple cellular pathways to correct lysosomal abnormalities in PD [253, 256,257,258,259]. Ambroxol also reduces ER stress in vivo, restores mitochondrial complex-I activity, reduces dopaminergic neuron loss, and improves behavioral symptoms in Drosophila and rodent models [185, 260, 261]. A clinical trial of ambroxol (ClinicalTrials.gov ID: NCT02941822) has demonstrated good cerebrospinal fluid penetration, safety, and tolerability while improving motor scores in patients [253]. These encouraging results paved the way for a phase 3 trial of ambroxol for the treatment of GBA1-PD, which will commence enrollment in the UK.
GBA1-targeting therapy
Gene therapies entail rectifying disease-related genetic abnormalities by introducing therapeutic genes and their accompanying regulatory components into the cell nucleus. In the context of GBA1-PD, gene therapies often utilize viral vectors carrying intact GBA1 gene sequences (Fig. 6). Direct administration or systemic injection of a functioning GBA1 gene into animals increased GCase activity, decreased GlcSph accumulation, and reduced α-syn accumulation, thereby improving motor and memory deficits and protecting dopaminergic neurons [24, 262]. Based on this promising evidence, a clinical trial of AAV9 (adeno-associated virus 9)-based gene therapy (LY3884961, formerly known as PR001) is currently underway for GBA1-PD. This phase 1/2a clinical trial involves administering multiple dosages of LY3884961 to evaluate its safety, tolerability, immunogenicity, biomarkers, and clinical effectiveness.

Gene therapies for GBA1-PD. Using the adeno-associated virus (AAV) as a vector, the normal GBA1 gene is introduced into the cells of patients through intracisternal injection to correct the defective gene and produce functional glucocerebrosidase (GCase) proteins, which in turn alleviates the disease
Since there are different classifications of GBA1 variants, pharmacological studies targeting GBA1-PD should carefully consider the differences between the various GBA1 variants to ensure that treatment is consistent with the mechanism of mutation and specificity of the GBA1 variant. Different GBA1 variants may result in diverse structures and functions of GCase. Certain variants may induce ER retention, preventing GCase from reaching the lysosome or causing ER stress and cellular demise [15, 184]; truncating mutations result in protein deletion and thus loss of function [23]; and others may affect enzyme function by altering the active site conformation [263]. Therefore, a balanced distribution of different mutations is essential during selection of participants to minimize the mutation-related bias. In addition, close attention should be paid to GCase modifiers such as CTSB, TMEM175, and LRRK2 when enrolling participants [65, 170, 237].
Conclusions
There is growing evidence underscoring the importance of GBA1 mutations in PD etiology. Research investigating the genotype-phenotype characteristics, pathogenesis, biomarkers, and targeted treatments for GBA1-PD has yielded significant insights. However, current understanding of the biological effects of various mutations remains incomplete. It is difficult to fully unravel the complexity of neurodegenerative diseases in cellular and animal models, so it is crucial to select appropriate GBA1-PD models according to the research objectives. It is believed that with the development of iPSCs, midbrain organoids, endolysosomal models, genome editing, optogenetics, and other technologies, more complete technical support will be provided to explore the mechanism of GBA1-PD [190, 213, 217, 264,265,266]. The treatment of GBA1-PD should be individualized, taking into account the GBA1 mutation status, other genetic factors, and comprehensive clinical features [244]. Because each therapeutic strategy has specific strengths and limitations, it is necessary to combine multiple therapeutic strategies to combat PD [267]. It is believed that with the emergence of novel therapies and technologies, such as GCase modulators, cell treatment, antisense oligonucleotides, microRNAs, ncRNAs, and CRISPR-Cas9, among others, there will be new opportunities for the treatment of GBA1-PD [38, 107, 110, 252, 268,269,270,271]. Finally, with the accumulation of cohort data, the establishment of standardized experimental procedures, the development of global cooperation, the advancement of detection technology, and the improvement of diagnosis and treatment, advances will be made in the clinical phenotypes, etiological mechanisms, biomarkers, and interventions and treatments for GBA1-PD.
Availability of data and materials
Not applicable.
Abbreviations
- α-syn:
-
Alpha-synuclein
- AD:
-
Alzheimer’s disease
- ALP:
-
Autophagy-lysosomal pathway
- BBB:
-
Blood–brain barrier
- BIN1 :
-
Bridging integrator 1
- CBE:
-
Conduritol B epoxide
- CMA:
-
Chaperone-mediated autophagy
- DLB:
-
Dementia with Lewy bodies
- ER:
-
Endoplasmic reticulum
- ERT:
-
Enzyme replacement therapy
- GBA1 :
-
Glucocerebrosidase gene
- GCase:
-
Glucocerebrosidase
- GD:
-
Gaucher’s disease
- GlcCer:
-
Glucosylceramide
- GlcSph:
-
Glucosylsphingosine
- GSLs:
-
Glycosphingolipids
- IL:
-
Interleukin
- LBs:
-
Lewy bodies
- LRRK2 :
-
Leucine-rich repeat kinase 2
- LSD:
-
Lysosome storage disorder
- PBMC:
-
Peripheral blood mononuclear cell
- PD:
-
Parkinson’s disease
- RBD:
-
Rapid eye movement sleep behavior disorder
- ROS:
-
Reactive oxygen species
- SRT:
-
Substrate reduction therapy
- UPR:
-
Unfolded protein response
References
Morris HR, Spillantini MG, Sue CM, Williams-Gray CH. The pathogenesis of Parkinson’s disease. Lancet. 2024;403(10423):293–304.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19(2):170–8.
Ye H, Robak LA, Yu M, Cykowski M, Shulman JM. Genetics and pathogenesis of Parkinson’s syndrome. Annu Rev Pathol. 2023;18:95–121.
Rizig M, Bandres-Ciga S, Makarious MB, Ojo OO, Crea PW, Abiodun OV, et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: a genome-wide association study. Lancet Neurol. 2023;22(11):1015–25.
Kim JJ, Vitale D, Otani DV, Lian MM, Heilbron K, et al. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat Genet. 2024;56(1):27–36.
Foo JN, Chew EGY, Chung SJ, Peng R, Blauwendraat C, Nalls MA, et al. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: a genome-wide association study. JAMA Neurol. 2020;77(6):746–54.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Bandres-Ciga S, Saez-Atienzar S, Kim JJ, Makarious MB, Faghri F, Diez-Fairen M, et al. Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol. 2020;140(3):341–58.
Udayar V, Chen Y, Sidransky E, Jagasia R. Lysosomal dysfunction in neurodegeneration: emerging concepts and methods. Trends Neurosci. 2022;45(3):184–99.
Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 2017;140(12):3191–203.
Horowitz M, Braunstein H, Zimran A, Revel-Vilk S, Goker-Alpan O. Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv Drug Deliv Rev. 2022;187:114402.
Droby A, Thaler A, Mirelman A. Imaging markers in genetic forms of Parkinson’s disease. Brain Sci. 2023;13(8):1212.
Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372(9645):1263–71.
Avenali M, Cerri S, Ongari G, Ghezzi C, Pacchetti C, Tassorelli C, et al. Profiling the biochemical signature of GBA-related Parkinson’s disease in peripheral blood mononuclear cells. Mov Disord. 2021;36(5):1267–72.
Thomas R, Moloney EB, Macbain ZK, Hallett PJ, Isacson O. Fibroblasts from idiopathic Parkinson’s disease exhibit deficiency of lysosomal glucocerebrosidase activity associated with reduced levels of the trafficking receptor LIMP2. Mol Brain. 2021;14(1):16.
Reczek D, Schwake M, Schroder J, Hughes H, Blanz J, Jin X, et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131(4):770–83.
Liu Y, Li S, Wang S, Yang Q, Wu Z, Zhang M, et al. LIMP-2 enhances cancer stem-like cell properties by promoting autophagy-induced GSK3beta degradation in head and neck squamous cell carcinoma. Int J Oral Sci. 2023;15(1):24.
Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020;30(6):452–66.
Ecard J, Lian YL, Divoux S, Gouveia Z, Vigne E, Perez F, et al. Lysosomal membrane proteins LAMP1 and LIMP2 are segregated in the Golgi apparatus independently of their clathrin adaptor binding motif. Mol Biol Cell. 2024;35(3):ar42.
Tatti M, Motta M, Di Bartolomeo S, Cianfanelli V, Salvioli R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy. 2013;9(2):241–3.
Suner L, Delhommeau F. Gaucher’s disease. N Engl J Med. 2022;386(20):1932.
Chen Y, Sud N, Hettinghouse A, Liu CJ. Molecular regulations and therapeutic targets of Gaucher disease. Cytokine Growth Factor Rev. 2018;41:65–74.
Smith L, Mullin S, Schapira AHV. Insights into the structural biology of Gaucher disease. Exp Neurol. 2017;298(Pt B):180–90.
Menozzi E, Toffoli M, Schapira AHV. Targeting the GBA1 pathway to slow Parkinson disease: insights into clinical aspects, pathogenic mechanisms and new therapeutic avenues. Pharmacol Ther. 2023;246:108419.
Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14(1):36.
Gegg ME, Menozzi E, Schapira AHV. Glucocerebrosidase-associated Parkinson disease: pathogenic mechanisms and potential drug treatments. Neurobiol Dis. 2022;166:105663.
Munoz SS, Petersen D, Marlet FR, Kucukkose E, Galvagnion C. The interplay between glucocerebrosidase, alpha-synuclein and lipids in human models of Parkinson’s disease. Biophys Chem. 2021;273:106534.
Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015;72(2):201–8.
Hanss Z, Boussaad I, Jarazo J, Schwamborn JC, Kruger R. Quality control strategy for CRISPR-Cas9-based gene editing complicated by a pseudogene. Front Genet. 2019;10:1297.
Miyoshi K, Hagita H, Horiguchi T, Tanimura A, Noma T. Redefining GBA gene structure unveils the ability of Cap-independent, IRES-dependent gene regulation. Commun Biol. 2022;5(1):639.
Spataro N, Roca-Umbert A, Cervera-Carles L, Valles M, Anglada R, Pagonabarraga J, et al. Detection of genomic rearrangements from targeted resequencing data in Parkinson’s disease patients. Mov Disord. 2017;32(1):165–9.
Tayebi N, Lichtenberg J, Hertz E, Sidransky E. Is Gauchian genotyping of GBA1 variants reliable? medRxiv. 2023:2023.10.26.23297627. https://doi.org/10.1101/2023.10.26.23297627.
Toffoli M, Chen X, Sedlazeck FJ, Lee CY, Mullin S, Higgins A, et al. Comprehensive short and long read sequencing analysis for the Gaucher and Parkinson’s disease-associated GBA gene. Commun Biol. 2022;5(1):670.
Dardis A, Michelakakis H, Rozenfeld P, Fumic K, Wagner J, Pavan E, et al. Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1. Orphanet J Rare Dis. 2022;17(1):442.
Jezela-Stanek A, Kleinotiene G, Chwialkowska K, Tylki-Szymanska A. Do not miss the (genetic) diagnosis of Gaucher syndrome: a narrative review on diagnostic clues and management in severe prenatal and perinatal-lethal sporadic cases. J Clin Med. 2021;10(21):4890.
Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567–83.
Straniero L, Rimoldi V, Samarani M, Goldwurm S, Di Fonzo A, Kruger R, et al. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci Rep. 2017;7(1):12702.
Dos Santos JCC, Mano GBC, da Cunha Barreto-Vianna AR, Garcia TFM, de Vasconcelos AV, Sa CSG, et al. The molecular impact of glucosylceramidase beta 1 (Gba1) in Parkinson's disease: a new genetic state of the art. Mol Neurobiol. 2024;61(9):6754–70.
Woo EG, Tayebi N, Sidransky E. Next-generation sequencing analysis of GBA1: the challenge of detecting complex recombinant alleles. Front Genet. 2021;12:684067.
Pachchek S, Landoulsi Z, Pavelka L, Schulte C, Buena-Atienza E, Gross C, et al. Accurate long-read sequencing identified GBA1 as major risk factor in the Luxembourgish Parkinson’s study. NPJ Parkinsons Dis. 2023;9(1):156.
McKeran RO, Bradbury P, Taylor D, Stern G. Neurological involvement in type 1 (adult) Gaucher’s disease. J Neurol Neurosurg Psychiatry. 1985;48(2):172–5.
Rizig M, Bandres-Ciga S, Makarious MB, Ojo OO, Crea PW, Abiodun OV, et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: a genome-wide association study. Lancet Neurol. 2023;22:1015–25.
Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014;71(6):752–7.
Eblan MJ, Walker JM, Sidransky E. The glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2005;352(7):728–31 (author reply 728–31).
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61.
Migdalska-Richards A, Schapira AH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem. 2016;139(Suppl 1):77–90.
Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet. 2011;20(1):202–10.
Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. 2015;84(9):880–7.
Parlar SC, Grenn FP, Kim JJ, Baluwendraat C, Gan-Or Z. Classification of GBA1 variants in Parkinson’s disease: the GBA1-PD browser. Mov Disord. 2023;38(3):489–95.
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132(Pt 7):1783–94.
Zhang Y, Shu L, Sun Q, Zhou X, Pan H, Guo J, et al. Integrated Genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front Mol Neurosci. 2018;11:43.
Chen Y, Gu X, Ou R, Zhang L, Hou Y, Liu K, et al. Evaluating the role of SNCA, LRRK2, and GBA in Chinese patients with early-onset Parkinson’s disease. Mov Disord. 2020;35(11):2046–55.
Li N, Wang L, Zhang J, Tan EK, Li J, Peng J, et al. Whole-exome sequencing in early-onset Parkinson’s disease among ethnic Chinese. Neurobiol Aging. 2020;90:150 e5-150 e11.
Yu Z, Wang T, Xu J, Wang W, Wang G, Chen C, et al. Mutations in the glucocerebrosidase gene are responsible for Chinese patients with Parkinson’s disease. J Hum Genet. 2015;60(2):85–90.
Zhao Y, Qin L, Pan H, Liu Z, Jiang L, He Y, et al. The role of genetics in Parkinson’s disease: a large cohort study in Chinese mainland population. Brain. 2020;143(7):2220–34.
Pulkes T, Choubtum L, Chitphuk S, Thakkinstian A, Pongpakdee S, Kulkantrakorn K, et al. Glucocerebrosidase mutations in Thai patients with Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(9):986–91.
Sun QY, Guo JF, Wang L, Yu RH, Zuo X, Yao LY, et al. Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson’s disease in Chinese population. Mov Disord. 2010;25(8):1005–11.
Trinh J, Guella I, Farrer MJ. Disease penetrance of late-onset Parkinsonism: a meta-analysis. JAMA Neurol. 2014;71(12):1535–9.
Balestrino R, Tunesi S, Tesei S, Lopiano L, Zecchinelli AL, Goldwurm S. Penetrance of glucocerebrosidase (GBA) mutations in Parkinson’s disease: a Kin cohort study. Mov Disord. 2020;35(11):2111–4.
Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78(6):417–20.
Menozzi E, Schapira AHV, Blandini F, Avenali M. Who is at risk of Parkinson disease? Refining the preclinical phase of GBA1 and LRRK2 variant carriers: a clinical, biochemical, and imaging approach. Curr Neurol Neurosci Rep. 2023;23(4):121–30.
Ji S, Wang C, Qiao H, Gu Z, Gan-Or Z, Fon EA, et al. Decreased penetrance of Parkinson’s disease in elderly carriers of glucocerebrosidase gene L444P/R mutations: a community-based 10-year longitudinal study. Mov Disord. 2020;35(4):672–8.
Blauwendraat C, Reed X, Krohn L, Heilbron K, Bandres-Ciga S, Tan M, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain. 2020;143(1):234–48.
Blauwendraat C, Tayebi N, Woo EG, Lopez G, Fierro L, Toffoli M, et al. Polygenic Parkinson’s disease genetic risk score as risk modifier of Parkinsonism in Gaucher disease. Mov Disord. 2023;38(5):899–903.
Leonard H, Blauwendraat C, Krohn L, Faghri F, Iwaki H, Ferguson G, et al. Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson’s disease. J Med Genet. 2020;57(5):331–8.
Zhou Y, Wang Y, Wan J, Zhao Y, Pan H, Zeng Q, et al. Mutational spectrum and clinical features of GBA1 variants in a Chinese cohort with Parkinson’s disease. NPJ Parkinsons Dis. 2023;9(1):129.
Fereshtehnejad SM, Romenets SR, Anang JB, Latreille V, Gagnon JF, Postuma RB. New clinical subtypes of Parkinson disease and their longitudinal progression: a prospective cohort comparison with other phenotypes. JAMA Neurol. 2015;72(8):863–73.
Caminiti SP, Carli G, Avenali M, Blandini F, Perani D. Clinical and dopamine transporter imaging trajectories in a cohort of Parkinson’s disease patients with GBA mutations. Mov Disord. 2022;37(1):106–18.
Chen YP, Yu SH, Zhang GH, Hou YB, Gu XJ, Ou RW, et al. The mutation spectrum of Parkinson-disease-related genes in early-onset Parkinson’s disease in ethnic Chinese. Eur J Neurol. 2022;29(11):3218–28.
Maple-Grodem J, Dalen I, Tysnes OB, Macleod AD, Forsgren L, Counsell CE, et al. Association of GBA genotype with motor and functional decline in patients with newly diagnosed Parkinson disease. Neurology. 2021;96(7):e1036–44.
Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, et al. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2020;91(7):695–702.
Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, et al. A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat Disord. 2017;36:47–51.
Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, et al. GBA-related Parkinson’s disease: dissection of genotype-phenotype correlates in a large Italian cohort. Mov Disord. 2020;35(11):2106–11.
Mullin S, Beavan M, Bestwick J, McNeill A, Proukakis C, Cox T, et al. Evolution and clustering of prodromal Parkinsonian features in GBA1 carriers. Mov Disord. 2019;34(9):1365–73.
Roeben B, Liepelt-Scarfone I, Lerche S, Zimmermann M, Wurster I, Sunkel U, et al. Longitudinal cognitive decline characterizes the profile of non-PD-manifest GBA1 mutation carriers. NPJ Parkinsons Dis. 2024;10(1):88.
Liu SY, Zheng Z, Gu ZQ, Wang CD, Tang BS, Xu YM, et al. Prevalence of pre-diagnostic symptoms did not differ between LRRK2-related, GBA-related and idiopathic patients with Parkinson’s disease. Parkinsonism Relat Disord. 2018;57:72–6.
Gan-Or Z, Mirelman A, Postuma RB, Arnulf I, Bar-Shira A, Dauvilliers Y, et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann Clin Transl Neurol. 2015;2(9):941–5.
Omer N, Giladi N, Gurevich T, Bar-Shira A, Gana-Weisz M, Glinka T, et al. Glucocerebrosidase activity is not associated with Parkinson’s disease risk or severity. Mov Disord. 2022;37(1):190–5.
Huang J, Cheng Y, Li C, Shang H. Genetic heterogeneity on sleep disorders in Parkinson’s disease: a systematic review and meta-analysis. Transl Neurodegener. 2022;11(1):21.
Avenali M, Toffoli M, Mullin S, McNeil A, Hughes DA, Mehta A, et al. Evolution of prodromal Parkinsonian features in a cohort of GBA mutation-positive individuals: a 6-year longitudinal study. J Neurol Neurosurg Psychiatry. 2019;90(10):1091–7.
Morris R, Martini DN, Ramsey K, Kelly VE, Smulders K, Hiller A, et al. Cognition as a mediator for gait and balance impairments in GBA-related Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):78.
Ren J, Zhou G, Wang Y, Zhang R, Guo Z, Zhou H, et al. Association of GBA genotype with motor and cognitive decline in Chinese Parkinson’s disease patients. Front Aging Neurosci. 2023;15:1091919.
Olszewska DA, McCarthy A, Soto-Beasley AI, Walton RL, Magennis B, McLaughlin RL, et al. Association between glucocerebrosidase mutations and Parkinson’s disease in Ireland. Front Neurol. 2020;11:527.
Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77(3):276–80.
Jesus S, Huertas I, Bernal-Bernal I, Bonilla-Toribio M, Caceres-Redondo MT, Vargas-Gonzalez L, et al. GBA variants influence motor and non-motor features of Parkinson’s disease. PLoS ONE. 2016;11(12):e0167749.
Ren J, Zhan X, Zhou H, Guo Z, Xing Y, Yin H, et al. Comparing the effects of GBA variants and onset age on clinical features and progression in Parkinson’s disease. CNS Neurosci Ther. 2023;30:e14387.
Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. 2015;30(3):407–11.
Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol. 2016;80(5):674–85.
Seto-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord. 2012;27(3):393–9.
Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, et al. GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord. 2016;31(1):95–102.
Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013;70(6):727–35.
Shiner T, Mirelman A, Gana Weisz M, Bar-Shira A, Ash E, Cialic R, et al. High frequency of GBA gene mutations in dementia with Lewy bodies among Ashkenazi Jews. JAMA Neurol. 2016;73(12):1448–53.
Ortega RA, Wang C, Raymond D, Bryant N, Scherzer CR, Thaler A, et al. Association of dual LRRK2 G2019S and GBA variations with Parkinson disease progression. JAMA Netw Open. 2021;4(4): e215845.
Omer N, Giladi N, Gurevich T, Bar-Shira A, Gana-Weisz M, Goldstein O, et al. A Possible modifying effect of the G2019S mutation in the LRRK2 gene on GBA Parkinson’s disease. Mov Disord. 2020;35(7):1249–53.
Ysselstein D, Nguyen M, Young TJ, Severino A, Schwake M, Merchant K, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun. 2019;10(1):5570.
Pang SY, Lo RCN, Ho PW, Liu HF, Chang EES, Leung CT, et al. LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl Neurodegener. 2022;11(1):5.
Szwedo AA, Dalen I, Pedersen KF, Camacho M, Backstrom D, Forsgren L, et al. GBA and APOE impact cognitive decline in Parkinson’s disease: a 10-year population-based study. Mov Disord. 2022;37(5):1016–27.
Saha O, Melo de Farias AR, Pelletier A, Siedlecki-Wullich D, Landeira BS, Gadaut J, et al. The Alzheimer's disease risk gene BIN1 regulates activity-dependent gene expression in human-induced glutamatergic neurons. Mol Psychiatry. 2024.
Ponnusamy M, Wang S, Yuksel M, Hansen MT, Blazier DM, McMillan JD, et al. Loss of forebrain BIN1 attenuates hippocampal pathology and neuroinflammation in a tauopathy model. Brain. 2023;146(4):1561–79.
Sudwarts A, Ramesha S, Gao T, Ponnusamy M, Wang S, Hansen M, et al. BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol Neurodegener. 2022;17(1):33.
Gan-Or Z, Amshalom I, Bar-Shira A, Gana-Weisz M, Mirelman A, Marder K, et al. The Alzheimer disease BIN1 locus as a modifier of GBA-associated Parkinson disease. J Neurol. 2015;262(11):2443–7.
Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 2016;73(10):1217–24.
van der Lee SJ, van Steenoven I, van de Beek M, Tesi N, Jansen IE, van Schoor NM, et al. Genetics contributes to concomitant pathology and clinical presentation in dementia with Lewy bodies. J Alzheimers Dis. 2021;83(1):269–79.
Ren J, Zhang R, Pan C, Xu J, Sun H, Hua P, et al. Prevalence and genotype-phenotype correlations of GBA-related Parkinson disease in a large Chinese cohort. Eur J Neurol. 2022;29(4):1017–24.
Simuni T, Uribe L, Cho HR, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol. 2020;19(1):71–80.
Davidson BA, Hassan S, Garcia EJ, Tayebi N, Sidransky E. Exploring genetic modifiers of Gaucher disease: the next horizon. Hum Mutat. 2018;39(12):1739–51.
Kumar M, Srikanth MP, Deleidi M, Hallett PJ, Isacson O, Feldman RA. Acid ceramidase involved in pathogenic cascade leading to accumulation of alpha-synuclein in iPSC model of GBA1-associated Parkinson’s disease. Hum Mol Genet. 2023;32(11):1888–900.
Burbulla LF, Krainc D. The role of dopamine in the pathogenesis of GBA1-linked Parkinson’s disease. Neurobiol Dis. 2019;132:104545.
Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19.
Polissidis A, Koronaiou E, Nikolopoulou G, Viel C, Nikatou M, Bogiongko M, et al. A double-hit in vivo model of GBA viral microRNA-mediated downregulation and human alpha-synuclein overexpression demonstrates nigrostriatal degeneration. Neurobiol Dis. 2022;163:105612.
Maor G, Rapaport D, Horowitz M. The effect of mutant GBA1 on accumulation and aggregation of alpha-synuclein. Hum Mol Genet. 2019;28(11):1768–81.
Garcia-Sanz P, Aerts JMFG, Moratalla R. The role of cholesterol in alpha-synuclein and Lewy body pathology in GBA1 Parkinson’s disease. Mov Disord. 2021;36(5):1070–85.
Pajares M, Rojo AI, Manda G, Bosca L, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020;9(7):1687.
Henderson MX, Sedor S, McGeary I, Cornblath EJ, Peng C, Riddle DM, et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological alpha-synuclein insult. Neuron. 2020;105(5):822-836 e7.
Wong YC, Krainc D. alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1–13.
Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 2017;357(6357):1255–61.
Nguyen M, Krainc D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A. 2018;115(21):5576–81.
Chatterjee D, Krainc D. Mechanisms of glucocerebrosidase dysfunction in Parkinson’s disease. J Mol Biol. 2023;435(12):168023.
Rosety I, Zagare A, Saraiva C, Nickels S, Antony P, Almeida C, et al. Impaired neuron differentiation in GBA-associated Parkinson’s disease is linked to cell cycle defects in organoids. NPJ Parkinsons Dis. 2023;9(1):166.
Behl T, Kaur G, Fratila O, Buhas C, Judea-Pusta CT, Negrut N, et al. Cross-talks among GBA mutations, glucocerebrosidase, and alpha-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: a comprehensive review. Transl Neurodegener. 2021;10(1):4.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52.
Aflaki E, Westbroek W, Sidransky E. The complicated relationship between Gaucher disease and Parkinsonism: insights from a rare disease. Neuron. 2017;93(4):737–46.
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain. 2014;137(Pt 3):834–48.
Yap TL, Gruschus JM, Velayati A, Sidransky E, Lee JC. Saposin C protects glucocerebrosidase against alpha-synuclein inhibition. Biochemistry. 2013;52(41):7161–3.
Liu G, Chen M, Mi N, Yang W, Li X, Wang P, et al. Increased oligomerization and phosphorylation of alpha-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol Aging. 2015;36(9):2649–59.
von Linstow CU, Gan-Or Z, Brundin P. Precision medicine in Parkinson’s disease patients with LRRK2 and GBA risk variants—let’s get even more personal. Transl Neurodegener. 2020;9(1):39.
Pchelina SN, Nuzhnyi EP, Emelyanov AK, Boukina TM, Usenko TS, Nikolaev MA, et al. Increased plasma oligomeric alpha-synuclein in patients with lysosomal storage diseases. Neurosci Lett. 2014;583:188–93.
Gundner AL, Duran-Pacheco G, Zimmermann S, Ruf I, Moors T, Baumann K, et al. Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol Dis. 2019;121:205–13.
Gaubert S, Hourregue C, Mouton-Liger F, Millot P, Franco M, Amar-Bouaziz E, et al. Exploring the link between GBA1 mutations and Dementia with Lewy bodies, a mini-review. Neurosci Biobehav Rev. 2022;141:104856.
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120(5):641–9.
Leyns CEG, Prigent A, Beezhold B, Yao L, Hatcher NG, Tao P, et al. Glucocerebrosidase activity and lipid levels are related to protein pathologies in Parkinson’s disease. NPJ Parkinsons Dis. 2023;9(1):74.
Plotegher N, Bubacco L, Greggio E, Civiero L. Ceramides in Parkinson’s disease: from recent evidence to new hypotheses. Front Neurosci. 2019;13:330.
Batta G, Soltesz L, Kovacs T, Bozo T, Meszar Z, Kellermayer M, et al. Alterations in the properties of the cell membrane due to glycosphingolipid accumulation in a model of Gaucher disease. Sci Rep. 2018;8(1):157.
Lerche S, Schulte C, Wurster I, Machetanz G, Roeben B, Zimmermann M, et al. The Mutation matters: CSF profiles of GCase, sphingolipids, alpha-synuclein in PD(GBA). Mov Disord. 2021;36(5):1216–28.
Merrill AH Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 2011;111(10):6387–422.
Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J Neurosci. 2017;37(40):9617–31.
Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M, et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016;590(6):716–25.
Lansbury P. The sphingolipids clearly play a role in Parkinson’s disease, but nature has made it complicated. Mov Disord. 2022;37(10):1985–9.
den Heijer JM, Cullen VC, Pereira DR, Yavuz Y, de Kam ML, Grievink HW, et al. A biomarker study in patients with GBA1-Parkinson’s disease and healthy controls. Mov Disord. 2023;38(5):783–95.
Galvagnion C, Brown JW, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, et al. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc Natl Acad Sci U S A. 2016;113(26):7065–70.
O’Leary EI, Jiang Z, Strub MP, Lee JC. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated alpha-synuclein. J Biol Chem. 2018;293(28):11195–205.
Fredriksen K, Aivazidis S, Sharma K, Burbidge KJ, Pitcairn C, Zunke F, et al. Pathological alpha-syn aggregation is mediated by glycosphingolipid chain length and the physiological state of alpha-syn in vivo. Proc Natl Acad Sci U S A. 2021;118(50):e2108489118.
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97(1):92-107 e10.
Yap TL, Jiang Z, Heinrich F, Gruschus JM, Pfefferkorn CM, Barros M, et al. Structural features of membrane-bound glucocerebrosidase and alpha-synuclein probed by neutron reflectometry and fluorescence spectroscopy. J Biol Chem. 2015;290(2):744–54.
Yap TL, Velayati A, Sidransky E, Lee JC. Membrane-bound alpha-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol Genet Metab. 2013;108(1):56–64.
Cosden M, Jinn S, Yao L, Gretzula CA, Kandebo M, Toolan D, et al. A novel glucosylceramide synthase inhibitor attenuates alpha synuclein pathology and lysosomal dysfunction in preclinical models of synucleinopathy. Neurobiol Dis. 2021;159:105507.
Lizama BN, Chu CT. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol Aspects Med. 2021;82:100972.
Sanyal A, DeAndrade MP, Novis HS, Lin S, Chang J, Lengacher N, et al. Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov Disord. 2020;35(5):760–73.
Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–76.
Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. 2021;40(19):e108863.
Nechushtai L, Frenkel D, Pinkas-Kramarski R. Autophagy in Parkinson’s disease. Biomolecules. 2023;13(10):1435.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5.
Suzuki K, Iseki E, Katsuse O, Yamaguchi A, Katsuyama K, Aoki I, et al. Neuronal accumulation of alpha- and beta-synucleins in the brain of a GM2 gangliosidosis mouse model. NeuroReport. 2003;14(4):551–4.
Garcia-Sanz P, Orgaz L, Fuentes JM, Vicario C, Moratalla R. Cholesterol and multilamellar bodies: Lysosomal dysfunction in GBA-Parkinson disease. Autophagy. 2018;14(4):717–8.
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67(12):1464–72.
Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, et al. Mutant glucocerebrosidase impairs alpha-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv. 2022;8(6):eabm6393.
Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. 2011;69(6):940–53.
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29(43):13578–88.
Hull A, Atilano ML, Gergi L, Kinghorn KJ. Lysosomal storage, impaired autophagy and innate immunity in Gaucher and Parkinson’s diseases: insights for drug discovery. Philos Trans R Soc Lond B Biol Sci. 1899;2024(379):20220381.
Kinghorn KJ, Asghari AM, Castillo-Quan JI. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen Res. 2017;12(3):380–4.
Lunghi G, Carsana EV, Loberto N, Cioccarelli L, Prioni S, Mauri L, et al. beta-Glucocerebrosidase deficiency activates an aberrant lysosome-plasma membrane axis responsible for the onset of neurodegeneration. Cells. 2022;11(15):2343.
Gegg ME, Sweet L, Wang BH, Shihabuddin LS, Sardi SP, Schapira AH. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov Disord. 2015;30(8):1085–9.
Straniero L, Rimoldi V, Monfrini E, Bonvegna S, Melistaccio G, Lake J, et al. Role of lysosomal gene variants in modulating GBA-associated Parkinson’s disease risk. Mov Disord. 2022;37(6):1202–10.
Machtel R, Boros FA, Dobert JP, Arnold P, Zunke F. From lysosomal storage disorders to Parkinson’s disease—challenges and opportunities. J Mol Biol. 2023;435(12):167932.
Lim SY, Tan AH, Ahmad-Annuar A, Klein C, Tan LCS, Rosales RL, et al. Parkinson’s disease in the Western Pacific region. Lancet Neurol. 2019;18(9):865–79.
Abe T, Kuwahara T. Targeting of lysosomal pathway genes for Parkinson’s disease modification: insights from cellular and animal models. Front Neurol. 2021;12:681369.
Kia DA, Zhang D, Guelfi S, Manzoni C, Hubbard L, Reynolds RH, et al. Identification of candidate Parkinson disease genes by integrating genome-wide association study, expression, and epigenetic data sets. JAMA Neurol. 2021;78(4):464–72.
Zhao YW, Pan HX, Liu Z, Wang Y, Zeng Q, Fang ZH, et al. The association between lysosomal storage disorder genes and Parkinson’s disease: a large cohort study in Chinese mainland population. Front Aging Neurosci. 2021;13:749109.
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49(10):1511–6.
Krohn L, Ozturk TN, Vanderperre B, Ouled Amar Bencheikh B, Ruskey JA, Laurent SB, et al. Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann Neurol. 2020;87(1):139–53.
Hopfner F, Mueller SH, Szymczak S, Junge O, Tittmann L, May S, et al. Rare variants in specific lysosomal genes are associated with Parkinson’s disease. Mov Disord. 2020;35(7):1245–8.
Tayebi N, Lopez G, Do J, Sidransky E, Pro-cathepsin D. Prosaposin, and progranulin: lysosomal networks in Parkinsonism. Trends Mol Med. 2020;26(10):913–23.
Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lullmann-Rauch R, et al. LIMP-2 expression is critical for beta-glucocerebrosidase activity and alpha-synuclein clearance. Proc Natl Acad Sci U S A. 2014;111(43):15573–8.
Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M, et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2015;10:15.
van Dijk KD, Persichetti E, Chiasserini D, Eusebi P, Beccari T, Calabresi P, et al. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov Disord. 2013;28(6):747–54.
Parnetti L, Chiasserini D, Persichetti E, Eusebi P, Varghese S, Qureshi MM, et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov Disord. 2014;29(8):1019–27.
Parnetti L, Paciotti S, Eusebi P, Dardis A, Zampieri S, Chiasserini D, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov Disord. 2017;32(10):1423–31.
Oftedal L, Maple-Grodem J, Dalen I, Tysnes OB, Pedersen KF, Alves G, et al. Association of CSF glucocerebrosidase activity with the risk of incident Dementia in patients with Parkinson disease. Neurology. 2023;100(4):e388–95.
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35(3):385–98.
Smith L, Schapira AHV. GBA variants and Parkinson disease: mechanisms and treatments. Cells. 2022;11(8):1261.
Maor G, Cabasso O, Krivoruk O, Rodriguez J, Steller H, Segal D, et al. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum Mol Genet. 2016;25(13):2712–27.
Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014;5:4028.
Kilpatrick BS, Magalhaes J, Beavan MS, McNeill A, Gegg ME, Cleeter MW, et al. Endoplasmic reticulum and lysosomal Ca(2)(+) stores are remodelled in GBA1-linked Parkinson disease patient fibroblasts. Cell Calcium. 2016;59(1):12–20.
Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, et al. ER stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 2016;6(3):342–56.
Sanchez-Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep. 2016;6:31380.
Schondorf DC, Ivanyuk D, Baden P, Sanchez-Martinez A, De Cicco S, Yu C, et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and Fly models of Parkinson’s disease. Cell Rep. 2018;23(10):2976–88.
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol. 2012;72(3):455–63.
Wright R. Mitochondrial dysfunction and Parkinson’s disease. Nat Neurosci. 2022;25(1):2.
Kim S, Wong YC, Gao F, Krainc D. Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease. Nat Commun. 2021;12(1):1807.
Baden P, Perez MJ, Raji H, Bertoli F, Kalb S, Illescas M, et al. Glucocerebrosidase is imported into mitochondria and preserves complex I integrity and energy metabolism. Nat Commun. 2023;14(1):1930.
Rubilar JC, Outeiro TF, Klein AD. The lysosomal beta-glucocerebrosidase strikes mitochondria: implications for Parkinson’s therapeutics. Brain. 2024;147:2610–20.
Klein AD, Outeiro TF. Glucocerebrosidase mutations disrupt the lysosome and now the mitochondria. Nat Commun. 2023;14(1):6383.
Cleeter MW, Chau KY, Gluck C, Mehta A, Hughes DA, Duchen M, et al. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int. 2013;62(1):1–7.
de la Mata M, Cotan D, Oropesa-Avila M, Garrido-Maraver J, Cordero MD, Villanueva Paz M, et al. Pharmacological chaperones and coenzyme Q10 treatment improves mutant beta-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci Rep. 2015;5:10903.
Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson’s disease. Cell Metab. 2013;17(6):941–53.
Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15(1):113–30.
Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200–10.
Malpartida AB, Williamson M, Narendra DP, Wade-Martins R, Ryan BJ. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy. Trends Biochem Sci. 2021;46(4):329–43.
Xu YH, Xu K, Sun Y, Liou B, Quinn B, Li RH, et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum Mol Genet. 2014;23(15):3943–57.
Munoz-Delgado L, Macias-Garcia D, Perinan MT, Jesus S, Adarmes-Gomez AD, Bonilla Toribio M, et al. Peripheral inflammatory immune response differs among sporadic and familial Parkinson’s disease. NPJ Parkinsons Dis. 2023;9(1):12.
Pandey MK, Burrow TA, Rani R, Martin LJ, Witte D, Setchell KD, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature. 2017;543(7643):108–12.
Boddupalli CS, Nair S, Belinsky G, Gans J, Teeple E, Nguyen TH, et al. Neuroinflammation in neuronopathic Gaucher disease: role of microglia and NK cells, biomarkers, and response to substrate reduction therapy. Elife. 2022;11:e79830.
Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4(1):27.
Rocha EM, Smith GA, Park E, Cao H, Graham AR, Brown E, et al. Sustained systemic glucocerebrosidase inhibition induces brain alpha-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid Redox Signal. 2015;23(6):550–64.
Mus L, Siani F, Giuliano C, Ghezzi C, Cerri S, Blandini F. Development and biochemical characterization of a mouse model of Parkinson’s disease bearing defective glucocerebrosidase activity. Neurobiol Dis. 2019;124:289–96.
Usenko T, Bezrukova A, Rudenok MM, Basharova K, Shadrina MI, Slominsky PA, et al. Whole transcriptome analysis of substantia Nigra in mice with MPTP-induced parkinsonism bearing defective glucocerebrosidase activity. Int J Mol Sci. 2023;24(15):12164.
Miliukhina IV, Usenko TS, Senkevich KA, Nikolaev MA, Timofeeva AA, Agapova EA, et al. Plasma cytokines profile in patients with Parkinson’s disease associated with mutations in GBA gene. Bull Exp Biol Med. 2020;168(4):423–6.
Chahine LM, Qiang J, Ashbridge E, Minger J, Yearout D, Horn S, et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 2013;70(7):852–8.
Aflaki E, Moaven N, Borger DK, Lopez G, Westbroek W, Chae JJ, et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell. 2016;15(1):77–88.
Bo RX, Li YY, Zhou TT, Chen NH, Yuan YH. The neuroinflammatory role of glucocerebrosidase in Parkinson’s disease. Neuropharmacology. 2022;207:108964.
Pitcairn C, Wani WY, Mazzulli JR. Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol Dis. 2019;122:72–82.
Kim J, Daadi EW, Oh T, Daadi ES, Daadi MM. Human induced pluripotent stem cell phenotyping and preclinical modeling of familial Parkinson’s disease. Genes (Basel). 2022;13(11):1937.
Yarkova ES, Grigor’eva EV, Medvedev SP, Pavlova SV, Zakian SM, Malakhova AA. IPSC-derived astrocytes contribute to in vitro modeling of Parkinson’s disease caused by the GBA1 N370S mutation. Int J Mol Sci. 2023;25(1):327.
Eichmuller OL, Knoblich JA. Human cerebral organoids—a new tool for clinical neurology research. Nat Rev Neurol. 2022;18(11):661–80.
Zagare A, Barmpa K, Smajic S, Smits LM, Grzyb K, Grunewald A, et al. Midbrain organoids mimic early embryonic neurodevelopment and recapitulate LRRK2-p.Gly2019Ser-associated gene expression. Am J Hum Genet. 2022;109(2):311–27.
Yahya V, Di Fonzo A, Monfrini E. Genetic evidence for endolysosomal dysfunction in Parkinson’s disease: a critical overview. Int J Mol Sci. 2023;24(7):6338.
Sanchiz-Calvo M, Bentea E, Baekelandt V. Rodent models based on endolysosomal genes involved in Parkinson’s disease. Curr Opin Neurobiol. 2022;72:55–62.
Migdalska-Richards A, Wegrzynowicz M, Rusconi R, Deangeli G, Di Monte DA, Spillantini MG, et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain. 2017;140(10):2706–21.
Migdalska-Richards A, Wegrzynowicz M, Harrison IF, Verona G, Bellotti V, Spillantini MG, et al. L444P Gba1 mutation increases formation and spread of alpha-synuclein deposits in mice injected with mouse alpha-synuclein pre-formed fibrils. PLoS ONE. 2020;15(8):e0238075.
Zhao X, Lin Y, Liou B, Fu W, Jian J, Fannie V, et al. PGRN deficiency exacerbates, whereas a brain penetrant PGRN derivative protects, GBA1 mutation-associated pathologies and diseases. Proc Natl Acad Sci U S A. 2023;120(1):e2210442120.
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138(Pt 9):2648–58.
Huh YE, Chiang MSR, Locascio JJ, Liao Z, Liu G, Choudhury K, et al. beta-Glucocerebrosidase activity in GBA-linked Parkinson disease: the type of mutation matters. Neurology. 2020;95(6):e685–96.
Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Crit Rev Oncog. 2013;18(3):163–75.
Atashrazm F, Hammond D, Perera G, Dobson-Stone C, Mueller N, Pickford R, et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci Rep. 2018;8(1):15446.
Kedariti M, Frattini E, Baden P, Cogo S, Civiero L, Ziviani E, et al. LRRK2 kinase activity regulates GCase level and enzymatic activity differently depending on cell type in Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):92.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406.
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–39.
Te Vruchte D, Sturchio A, Priestman DA, Tsitsi P, Hertz E, Andreasson M, et al. Glycosphingolipid changes in plasma in Parkinson’s disease independent of glucosylceramide levels. Mov Disord. 2022;37(10):2129–34.
Huebecker M, Moloney EB, van der Spoel AC, Priestman DA, Isacson O, Hallett PJ, et al. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener. 2019;14(1):40.
Surface M, Balwani M, Waters C, Haimovich A, Gan-Or Z, Marder KS, et al. Plasma glucosylsphingosine in GBA1 mutation carriers with and without Parkinson’s disease. Mov Disord. 2022;37(2):416–21.
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, et al. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol. 2015;2(4):433–8.
Blumenreich S, Nehushtan T, Barav OB, Saville JT, Dingjan T, Hardy J, et al. Elevation of gangliosides in four brain regions from Parkinson’s disease patients with a GBA mutation. NPJ Parkinsons Dis. 2022;8(1):99.
Huh YE, Park H, Chiang MSR, Tuncali I, Liu G, Locascio JJ, et al. Glucosylceramide in cerebrospinal fluid of patients with GBA-associated and idiopathic Parkinson’s disease enrolled in PPMI. NPJ Parkinsons Dis. 2021;7(1):102.
Ysselstein D, Young TJ, Nguyen M, Padmanabhan S, Hirst WD, Dzamko N, et al. Evaluation of strategies for measuring lysosomal glucocerebrosidase activity. Mov Disord. 2021;36(12):2719–30.
Oftedal L, Maple-Grodem J, Forland MGG, Alves G, Lange J. Validation and assessment of preanalytical factors of a fluorometric in vitro assay for glucocerebrosidase activity in human cerebrospinal fluid. Sci Rep. 2020;10(1):22098.
Farfel-Becker T, Do J, Tayebi N, Sidransky E. Can GBA1-associated Parkinson disease be modeled in the mouse? Trends Neurosci. 2019;42(9):631–43.
Sosero YL, Yu E, Krohn L, Rudakou U, Mufti K, Ruskey JA, et al. LRRK2 p.M1646T is associated with glucocerebrosidase activity and with Parkinson’s disease. Neurobiol Aging. 2021;103:142 e1-142 e5.
Cecioni S, Ashmus RA, Gilormini PA, Zhu S, Chen X, Shan X, et al. Quantifying lysosomal glycosidase activity within cells using bis-acetal substrates. Nat Chem Biol. 2022;18(3):332–41.
Senkevich K, Rudakou U, Gan-Or Z. New therapeutic approaches to Parkinson’s disease targeting GBA, LRRK2 and Parkin. Neuropharmacology. 2022;202:108822.
Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A. 2017;114(10):2699–704.
Viel C, Clarke J, Kayatekin C, Richards AM, Chiang MSR, Park H, et al. Preclinical pharmacology of glucosylceramide synthase inhibitor venglustat in a GBA-related synucleinopathy model. Sci Rep. 2021;11(1):20945.
Giladi N, Alcalay RN, Cutter G, Gasser T, Gurevich T, Hoglinger GU, et al. Safety and efficacy of venglustat in GBA1-associated Parkinson’s disease: an international, multicentre, double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2023;22(8):661–71.
Peterschmitt MJ, Saiki H, Hatano T, Gasser T, Isaacson SH, Gaemers SJM, et al. Safety, pharmacokinetics, and pharmacodynamics of oral venglustat in patients with Parkinson’s disease and a GBA mutation: results from part 1 of the randomized, double-blinded, placebo-controlled MOVES-PD trial. J Parkinsons Dis. 2022;12(2):557–70.
Huh YE, Usnich T, Scherzer CR, Klein C, Chung SJ. GBA1 variants and Parkinson’s disease: paving the way for targeted therapy. J Mov Disord. 2023;16(3):261–78.
Zimran A, Revel-Vilk S, Becker-Cohen M, Istaiti M, Rolfs A. Venglustat in GBA1-related Parkinson’s disease. Lancet Neurol. 2024;23(2):137.
Istaiti M, Revel-Vilk S, Becker-Cohen M, Dinur T, Ramaswami U, Castillo-Garcia D, et al. Upgrading the evidence for the use of ambroxol in Gaucher disease and GBA related Parkinson: investigator initiated registry based on real life data. Am J Hematol. 2021;96(5):545–51.
Santana AG, Robinson K, Vickers C, Deen MC, Chen HM, Zhou S, et al. Pharmacological chaperones for GCase that switch conformation with pH enhance enzyme levels in Gaucher animal models. Angew Chem Int Ed Engl. 2022;61(38):e202207974.
Zhang K, Zhu S, Li J, Jiang T, Feng L, Pei J, et al. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm Sin B. 2021;11(10):3015–34.
Martinez-Bailen M, Clemente F, Matassini C, Cardona F. GCase enhancers: a potential therapeutic option for gaucher disease and other neurological disorders. Pharmaceuticals (Basel). 2022;15(7):823.
Kopytova AE, Rychkov GN, Cheblokov AA, Grigor’eva EV, Nikolaev MA, Yarkova ES, et al. Potential binding sites of pharmacological chaperone NCGC00241607 on mutant beta-glucocerebrosidase and its efficacy on patient-derived cell cultures in gaucher and Parkinson’s disease. Int J Mol Sci. 2023;24(10):9105.
Han TU, Sam R, Sidransky E. Small molecule chaperones for the treatment of Gaucher disease and GBA1-associated Parkinson disease. Front Cell Dev Biol. 2020;8:271.
Burbulla LF, Jeon S, Zheng J, Song P, Silverman RB, Krainc D. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson's disease. Sci Transl Med. 2019;11(514):eaau6870.
Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 2020;77(4):427–34.
Yang SY, Taanman JW, Gegg M, Schapira AHV. Ambroxol reverses tau and alpha-synuclein accumulation in a cholinergic N370S GBA1 mutation model. Hum Mol Genet. 2022;31(14):2396–405.
Siemeling O, Slingerland S, van der Zee S, van Laar T. Study protocol of the GRoningen early-PD Ambroxol treatment (GREAT) trial: a randomized, double-blind, placebo-controlled, single center trial with ambroxol in Parkinson patients with a GBA mutation. BMC Neurol. 2024;24(1):146.
Cyske Z, Gaffke L, Rintz E, Wisniewska K, Wegrzyn G, Pierzynowska K. Molecular mechanisms of the ambroxol action in Gaucher disease and GBA1 mutation-associated Parkinson disease. Neurochem Int. 2024;178:105774.
McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137(Pt 5):1481–95.
Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis. 2015;82:235–42.
Magalhaes J, Gegg ME, Migdalska-Richards A, Schapira AH. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci Rep. 2018;8(1):1385.
Mishra A, Krishnamurthy S. Neurorestorative effects of sub-chronic administration of ambroxol in rodent model of Parkinson’s disease. Naunyn Schmiedebergs Arch Pharmacol. 2020;393(3):429–44.
Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol. 2016;80(5):766–75.
Vieira SRL, Schapira AHV. Glucocerebrosidase mutations and Parkinson disease. J Neural Transm (Vienna). 2022;129(9):1105–17.
Toffoli M, Smith L, Schapira AHV. The biochemical basis of interactions between glucocerebrosidase and alpha-synuclein in GBA1 mutation carriers. J Neurochem. 2020;154(1):11–24.
Okano H, Morimoto S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell. 2022;29(2):189–208.
Mansour HM, El-Khatib AS. Exploring Parkinson-associated kinases for CRISPR/Cas9-based gene editing: beyond alpha-synuclein. Ageing Res Rev. 2023;92:102114.
Kim MS, Ra EA, Kweon SH, Seo BA, Ko HS, Oh Y, et al. Advanced human iPSC-based preclinical model for Parkinson’s disease with optogenetic alpha-synuclein aggregation. Cell Stem Cell. 2023;30(7):973-986 e11.
Kim MJ, Kim S, Reinheckel T, Krainc D. Inhibition of cysteine protease cathepsin Lincreases the level and activity of lysosomal glucocerebrosidase. JCI Insight. 2024;9(3).
Gehrlein A, Udayar V, Anastasi N, Morella ML, Ruf I, Brugger D, et al. Targeting neuronal lysosomal dysfunction caused by beta-glucocerebrosidase deficiency with an enzyme-based brain shuttle construct. Nat Commun. 2023;14(1):2057.
Chen C, Hertz E, Chen Y, Sidransky E. Targeting protein clearance pathways in GBA1-associated Parkinson disease. Expert Opin Ther Targets. 2022;26(12):1031–5.
Ryan E, Seehra G, Sharma P, Sidransky E. GBA1-associated Parkinsonism: new insights and therapeutic opportunities. Curr Opin Neurol. 2019;32(4):589–96.
Peng Y, Liou B, Lin Y, Mayhew CN, Fleming SM, Sun Y. iPSC-derived neural precursor cells engineering GBA1 recovers acid beta-glucosidase deficiency and diminishes alpha-synuclein and neuropathology. Mol Ther Methods Clin Dev. 2023;29:185–201.
Jo J, Yang L, Tran HD, Yu W, Sun AX, Chang YY, et al. Lewy body-like inclusions in human midbrain organoids carrying glucocerebrosidase and alpha-synuclein mutations. Ann Neurol. 2021;90(3):490–505.
Gegg ME, Verona G, Schapira AHV. Glucocerebrosidase deficiency promotes release of alpha-synuclein fibrils from cultured neurons. Hum Mol Genet. 2020;29(10):1716–28.
Baden P, Yu C, Deleidi M. Insights into GBA Parkinson’s disease pathology and therapy with induced pluripotent stem cell model systems. Neurobiol Dis. 2019;127:1–12.
Yang W, Li X, Yin N. Increased alpha-synuclein oligomerization is associated with decreased activity of glucocerebrosidase in the aging human striatum and hippocampus. Neurosci Lett. 2020;733:135093.
Xicoy H, Wieringa B, Martens GJ. The SH-SY5Y cell line in Parkinson’s disease research: a systematic review. Mol Neurodegener. 2017;12(1):10.
Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun. 2014;5:4755.
Bae EJ, Yang NY, Lee C, Lee HJ, Kim S, Sardi SP, et al. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp Mol Med. 2015;47(3):e153.
Fishbein I, Kuo YM, Giasson BI, Nussbaum RL. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain. 2014;137(Pt 12):3235–47.
Papadopoulos VE, Nikolopoulou G, Antoniadou I, Karachaliou A, Arianoglou G, Emmanouilidou E, et al. Modulation of beta-glucocerebrosidase increases alpha-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum Mol Genet. 2018;27(10):1696–710.
Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A. 2013;110(9):3537–42.
Deng YN, Shi J, Liu J, Qu QM. Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem Int. 2013;63(1):1–9.
Acknowledgements
The authors thank the funding sources for supporting this work.
Funding
This work was supported by the National Key Plan for Scientific Research and Development of China (2021YFC2501204), the National Key R&D Program of China (2021YFC2502100), the National Natural Science Foundation of China (81873785, 82071439, 81974202, and U20A20355), Technology Major Project of Hunan Provincial Science and Technology Department (2021SK1010), the Innovation-driven Team Project from Central South University (2020CX016), and the Innovative Team Program from the Department of Science & Technology of Hunan Province (2019RS1010).
Author information
Authors and Affiliations
Contributions
XZ critically reviewed the pieces of literature and wrote the manuscript. HW and BT edited and revised important points. JG took responsibility for all aspects of the work and ensured that issues related to the accuracy or integrity of any part of the work were properly investigated and resolved. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Supplementary Information
Additional file 1: Table S1
GBA1 variants reported in PD.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, X., Wu, H., Tang, B. et al. Clinical, mechanistic, biomarker, and therapeutic advances in GBA1-associated Parkinson’s disease. Transl Neurodegener 13, 48 (2024). https://doi.org/10.1186/s40035-024-00437-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-024-00437-6