
Abstract
Background
Current clinical diagnosis pathway for lysosomal storage disorders (LSDs) involves sequential biochemical enzymatic tests followed by DNA sequencing, which is iterative, has low diagnostic yield and is costly due to overlapping clinical presentations. Here, we describe a novel low-cost and high-throughput sequencing assay using single-molecule molecular inversion probes (smMIPs) to screen for causative single nucleotide variants (SNVs) and copy number variants (CNVs) in genes associated with 29 common LSDs in India.
Results
903 smMIPs were designed to target exon and exon–intron boundaries of targeted genes (n = 23; 53.7 kb of the human genome) and were equimolarly pooled to create a sequencing library. After extensive validation in a cohort of 50 patients, we screened 300 patients with either biochemical diagnosis (n = 187) or clinical suspicion (n = 113) of LSDs. A diagnostic yield of 83.4% was observed in patients with prior biochemical diagnosis of LSD. Furthermore, diagnostic yield of 73.9% (n = 54/73) was observed in patients with high clinical suspicion of LSD in contrast with 2.4% (n = 1/40) in patients with low clinical suspicion of LSD. In addition to detecting SNVs, the assay could detect single and multi-exon copy number variants with high confidence. Critically, Niemann-Pick disease type C and neuronal ceroid lipofuscinosis-6 diseases for which biochemical testing is unavailable, could be diagnosed using our assay. Lastly, we observed a non-inferior performance of the assay in DNA extracted from dried blood spots in comparison with whole blood.
Conclusion
We developed a flexible and scalable assay to reliably detect genetic causes of 29 common LSDs in India. The assay consolidates the detection of multiple variant types in multiple sample types while having improved diagnostic yield at same or lower cost compared to current clinical paradigm.
Similar content being viewed by others


Background
Lysosomal storage disorders (LSDs) are a group of ~ 70 monogenic metabolic disorders caused due to defect in the genes encoding lysosomal proteins and is estimated to have a combined incidence 1 in 1500 to 7000 live births [1]. Genes associated with LSDs include acid hydrolases, integral membrane proteins, activators and transporters [2]. LSDs are characterized by unwanted accumulation of metabolic substrate inside lysosomes, leading to cellular dysfunction and/or cell death. Clinical symptoms in children with LSDs develop progressively over time resulting in a wide spectrum of manifestations with variable severity. Most LSDs manifest in early childhood, however, late-onset juvenile and adult forms have also been reported [2,3,4]. In view of the recent development of therapeutic strategies for LSDs like enzyme replacement therapy (ERT), stem cell transplantation and emerging gene therapy for many LSDs, early diagnosis of LSDs is of particular relevance [5, 6].
The present diagnostic approach for LSDs includes a primary clinical evaluation followed by biochemical screening, confirmatory enzyme tests based on the detection of accumulated substrates and genetic study [2]. This 3-step diagnostic pathway is considered as “gold standard” for LSD diagnosis. However, enzyme testing is not available for some LSD types like the Niemann pick type C, activator protein deficiency and some forms of neuronal ceroid lipofuscinosis [2]. Furthermore, the enzyme testing involves analysis of a single enzymatic reaction at a given time, therefore, the approach is iterative in nature, especially in cases whereby multiple LSDs have overlapping phenotypic presentation and require sequential rounds of enzymatic testing to identify the disease. Overall, this route is time-consuming and expensive leading to poor diagnostic yields and long time to diagnosis [7].
India has a significant burden of LSDs, as indicated by several independent groups [8]. There is a high prevalence of Gaucher disease followed by the mucopolysaccharidosis group and approximately 20 other LSDs [8]. However, there are several challenges in achieving an accurate and timely diagnosis of LSDs in India. One of the reasons being paucity of quality assured diagnostic labs for biochemical enzyme based assays in the country. Furthermore, a study by Agarwal et al. 2015 demonstrated that the median time to reach a final diagnosis after the disease onset was 14 months [7]. This diagnostic delay has prognostic as well as therapeutic implications. Hence, there is a significant scope and need for improvement in the diagnosis of LSDs in India.
Significant advancements in next generation sequencing (NGS) technologies has led to simultaneous investigation of multiple genes with high accuracy and reduced costs compared to traditional biochemical assays. Indeed, several studies have explored use of multigene NGS panels for genetic diagnosis of LSDs with improved diagnostic yield compared to biochemical assays [9,10,11]. However, these panels have low diagnostic yields for diseases whereby the causative gene has a high sequence similarity with its pseudogene or consists of low complexity sequence region [12, 13]. Furthermore, multiplex ligation probe dependent amplification (MLPA) is required to detect copy number variations (CNV) in cases whereby single nucleotide variants (SNVs) have been ruled out by NGS panels, thereby adding complexity and cost to the diagnostic pathway.
Previously, single molecule molecular inversion probe (smMIP) coupled with unique molecular barcode (UMB) based target capture protocol followed by NGS has been used to detect both germline and somatic SNVs, CNVs and indels with high accuracy [14,15,16]. The key characteristics of this technique include consensus variant calling through the use of UMB, low cost per sample, minimum input DNA requirement, and high flexibility to include and exclude genes in the target capture step as required [16]. The potential for this highly flexible and affordable methodology in clinical practice is underlined by its low per sample cost coupled with easily manageable and scalable protocol [16, 17]. This approach therefore is likely to aid in improving genetic diagnostics of LSDs, especially in low-middle income countries (LMICs) like India.
Therefore, we developed and validated smMIP based NGS assay targeting coding regions of 23 genes that are associated with 29 common LSDs in India [18,19,20]. Furthermore, we validated this assay for its use on germline DNA sample extracted from dried blood spots, in order to increase its utility in clinical settings whereby dried blood spot sample type is only available. We hypothesized that this approach could reverse the current clinical “gold standard” diagnostic algorithm for LSDs whereby, the smMIP-NGS assay could be used as a first-line genetic test in patients clinically suspected with one of the 29 common LSDs in India followed by a biochemical and enzyme test to confirm the molecular findings. This alternative approach could help in reducing the time to reach a diagnosis and help initiate treatment. In this paper, we validated the assay on positive control samples that had been diagnosed with currently used methods. In addition, we studied the diagnostic value of this assay in a cohort of 300 clinically suspected or enzymatically diagnosed patients with LSDs.
Methods
Gene selection
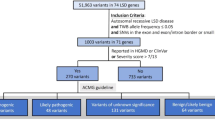
Previous studies by Sheth et al. 2014 and Verma et al. 2012 have addressed the burden of LSDs in India and identified the most common LSDs [19, 20]. The selection of genes followed the American College of Medical Genetics (ACMG) guidelines on gene panel design for diagnostic purposes and reporting [21]. Based on the disease prevalence estimates, we included genes for seven classes of LSDs-sphingolipidoses, mucopolysaccharidosis, neuronal ceroid lipofuscinoses, integral membrane protein disorders, post-translational modification defects, activator protein deficiency and glycogen storage disorder. A total set of 23 genes with known association with the shortlisted LSDs were selected (Table 1).
Patient cohort
Validation cohort: A total of 50 anonymized genomic DNA samples of patients with prior confirmed diagnosis with one of the 29 LSDs at FRIGE Institute of Human Genetics between 2008 and 2018 were obtained. All patients were diagnosed through biochemical and genetic tests. Genetic diagnosis was carried out using Sanger sequencing in 44 cases, MLPA in 4 cases and clinical exome sequencing in 2 cases.
Diagnostic yield cohort: We enrolled a total of 300 patients which were divided into two groups. The first group comprised of 187 patients which had only biochemical diagnosis for a given LSD. The second group comprised of 113 patients with a high clinical suspicion for one of the 29 common LSDs but no prior biochemical or genetic test was carried out. For all patients, genomic DNA was extracted from the peripheral blood sample of the patients by salting out protocol [22].
The ethics committee of the Foundation for Research in Genetics and Endocrinology (FRIGE) approved the study at the Institute of Human Genetics (Reg No- E/13237). The study comprised DNA samples of patients referred from Institute of Human Genetics, Ahmedabad as well as from other hospitals/reference laboratory/consultants across the country (Additional file 1). A written informed consent for the study was obtained from the guardians of all the participating subjects as per the Helsinki declaration.
DNA extraction from dried blood spot (DBS)
Three patient samples whereby genetic diagnosis of LSD was carried out using the smMIP based assay in the germline DNA from EDTA treated peripheral blood sample were selected. Fresh peripheral blood sample was spotted on the dried blood spot (DBS) cards (HiMedia, India). DNA extraction from DBS cards was carried out in accordance with the InstaDNA kit protocol (HiMedia, India), with minor modifications. Briefly, 12 card punches of 3 mm size each was taken. The initial procedure was carried out in a petri plate for even distribution of wash with 1.5 ml of distilled water for 5 min. This was followed by transferring of the punch cards to 1.5 ml Eppendorf tubes containing 10 µl of proteinase K and 300 µl of wash solution. The sample was incubated at 65 °C for 15 min in a shaking incubator to facilitate cell lysis and DNA release. The supernatant was discarded, and the punch cards were washed with 300 µl of wash solution. A second wash was performed using 400 µl of TE buffer to rinse traces of salts and ethanol and the supernatant was subsequently discarded. The punch cards were then transferred to a petri plate for uniform drying at 65 °C for 15 min. Once dried, the punch cards wete transferred to a sterile tube containing 50 µl of Solution ID1. After pulse vortexing, the tubes were incubated at 65 °C for 15 min. Finally, 100 µl of ID2 solution was added and the sample was incubated at room temperature for 10 min. Extracted genomic DNA sample in the supernatant was collected in a sterile tube which was stored at − 20 °C. All genomic DNA samples were quantified using QIAexpert (Qiagen, Germany) and Qubit (Themo Fisher Scientific, USA).
smMIP design
All smMIPs targeting the exons and intron–exon boundaries of the 23 genes were designed using the MIPGEN pipeline [23] and hg19/GRCh37 human reference genome build. The smMIP probe consisted of a 30 bp common linker arm containing a 5 bp random tag next to the extension arm. The random tag, also known as UMB, in the backbone of each MIP helps distinguish 1024 (45) unique genomic DNA equivalents. This helps reduce potential PCR errors through removal of PCR duplicates and result in high-quality reads that helps detect SNV and CNV with high accuracy. Each smMIP probe covered a 110 bp genomic region with a maximum and minimum overlap of 40 and 20 bp, respectively, with the adjacent smMIP. Combined, 903 smMIPs targeted approximately 53.7 kb of the human genome (Additional file 2).
smMIP pooling and phosphorylation
All 903 smMIP probes were pooled at a final concentration of 0.1 µM followed by phosphorylation. smMIPs were phosphorylated using 20U of T4 Polynucleotide Kinase (New England Biolabs, USA), 1X T4 DNA ligase buffer (New England Biolabs, USA), 50 µl of 0.1 µM of pooled smMIPs in a total reaction volume of 60 µl and incubated at 37֯ °C for 45 min followed by 65 °C for 20 min.
smMIP capture, library preparation and sequencing
100 ng and 20 ng of genomic DNA from whole blood and DBS sample, respectively, was quantified using a Qubit dsDNA HS assay kit (Thermo Fisher Scientific, USA). It was subjected to smMIP capture in accordance with the protocol previously described with minor modifications [16, 24]. One hundred nanogram of genomic DNA was used as input and the target genomic regions were captured in a reaction containing smMIPs to genomic DNA in a molecular ratio of 1000:1. The capturing conditions were 95 °C for 10 min for denaturation of the double-stranded template DNA followed by 17 h of incubation at 60 °C. During this period, phosphorylated smMIPs hybridized against the single-stranded DNA followed by a gap-fill reaction and ligation to form circularized probes. All non-circularized probes and residual unused template DNA were digested in the following exonuclease treatment step. For amplifying the resultant circularized targets, 2X iProof Master Mix (BioRad, USA), common forward primer, and sample barcoded reverse primers were used. The thermal cycling conditions were as follows: 30 s at 98 °C followed by 19 cycles of 98 °C for 15 s, 60 °C for 30 s and 72 °C for 30 s, followed by 72 °C for 2 min. The primers used during this step contained adaptors compatible with Illumina sequencing platforms (Illumina, USA) [16]. The smMIP amplification products (269 bp) were analyzed on a 2% agarose gel.
After PCR, all the barcoded individual patient libraries were pooled together in equal volumes and purified using Agencourt AMPure XP beads as per the manufacturer’s protocol (Beckman Coulter, USA). The pooled and purified library was diluted to a concentration of 4 nM in 10 mM Tris EDTA (pH 8.5) and sequenced on Illumina MiSeq platform (Illumina, USA) using custom sequencing primers and 2 × 156 bp paired-end reads [16].
Rebalancing the smMIP pool
In order to reduce the sequence coverage variability observed in the initial MIP experiment, the smMIP pool was rebalanced by adding a higher concentration (10x) of the underperforming smMIPs and an equimolar concentration of the unphosphorylated probes of overperforming smMIPs. The final concentration of each smMIP is provided in Additional file 2.
Data analysis pipeline
All the FASTQ files containing the forward and reverse reads from all the samples were processed by trimming the 5 bp random tag from the reads and kept in key identifiers for later use. Following this, the reads were aligned to the hg19/GRCh37 human reference genome using BWA-MEM (v.0.7.12) [25] with the output presented as a sample specific BAM file amalgamated with the UMB data. Reads from the same smMIP i.e., containing the same UMB were discarded at random from the BAM file and the final coverage for individual smMIP was written to a coverage report. Single Nucleotide Variants (SNVs) were called using the GATK HaplotypeCaller (v4.1.2) following base quality score recalibration step, in accordance with the GATK best practice guidelines [26]. Variants were annotated, filtered and prioritized based on the patient’s phenotype (in HPO format) using Exomiser v12 [27] integrating data from SIFT (https://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html), Polyphen2 (http://genetics.bwh.harward.edu/pph2), MutationTaster (http://www.mutationtaster.org), Combined Annotation Dependent Depletion (CADD) scores, REVEL scores, dbSNP (www.ncbi.nlm.nih.gov/SNP/), the Genome Aggregation Database (gnomAD; gnomad.broadinstitute.org) and ClinVar (www.ncbi.nlm.nih.gov/clinvar).
For detection of SNVs, BAM files of all the samples processed in a single sequencing run were used to normalize the coverage data and detect upto single-exon level CNVs. The CNV calls were carried out using DECoN v1.0.1 [28] using a minimum of 17 samples per batch for analysis.
Variant validation and classification
Candidate variants identified in the patient samples were prioritized based on the minor allele frequency in the public databases, correlation of patient phenotype and biochemical report if available, predicted protein impact and predicted pathogenicity scores. All identified variants were assessed using Integrative Genomics Viewer (IGV) version 2.12.3 for read depth and read bias. Candidate SNVs were validated using specifically designed primers (https://bioinfo.ut.ee/primer3-0.4.0/) followed by Sanger sequencing for confirmation. The PCR products were purified using Exo-SAP-IT™ (USB Corporation, USA) and subjected to di-deoxy chain termination protocol using BigDye Terminator v3.1 cycle sequencing kit (Thermo Fisher Scientific, USA) and capillary electrophoresis was performed using an automated sequencer SeqStudio (Applied Biosystem, USA). Sequences were assessed by comparing with the hg19/GRCh37 genomic reference sequence of the specific genes using NCBI-BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). All candidate CNVs were validated using SYBR green dye (KAPA Biosystems, USA) based Q-PCR with ROX dye as a passive reference dye. Q-PCR reaction was carried out on the StepOne thermal cycler (Applied Biosystems, USA). Briefly, the reference gene used for the Q-PCR assay was ALB. Relative quantification approach was utilized whereby the Ct value was used as a determinant of the differences in the number of copies of the target sequence in different samples. Relative quantification (RQ) value of 0.5, 1 and 1.5 was suggestive of 1, 2 and 3 copies of the target sequence, respectively.
Finally, pathogenicity of the variants was classified according to the ACMG-AMP guidelines and ClinGen framework [29,30,31].
Whole exome sequencing
Genomic DNA of 24 patients in whom no genetic diagnosis could be made using smMIP based assay was subjected to selective capture and sequencing of the protein coding regions using Human Core Exome enrichment kit (Twist Biosciences, USA). The prepared library was subjected to paired-end sequencing with a mean coverage of > 80-100X on the Illumina NovaSeq 6000 platform (Illumina, USA). FASTQ files were aligned against human reference genome GRCh37/hg19 using BWA MEM v0.7.12 [25]. SNVs and indels were called using GATK v4.12 HaplotypeCaller [26]. Additionally, CNVs were called using ExomeDepth v1.1.10 [32]. Variant annotation, filtration and prioritization was carried out using Exomiser v12.1.0 [27].
Whole genome sequencing
Whole genome sequencing was performed for 7 cases at the Yale Centre for Genome Analysis whereby the cases were diagnosed with one of the 29 LSDs through biochemical assay but no genetic cause was identified in the smMIP based assay. 0.5ug of genomic DNA was enzymatically fragmented and end-repaired in a single reaction using xGen™ DNA EZ Library Prep Kit (IDT, USA). Size of the final library construct was determined on Caliper LabChip GXsystem and quantification was performed by Q-PCR SYBR Green reactions with a set of DNA standards using the Kapa Library Quantification Kit (KAPA Biosystems, USA). Libraries were sequenced on the Illumina NovaSeq 6000 platform and S4 flow cells with 2 × 150 bp paired-end reads and with yield of at least 700Gbp passing filter data per lane. Sample demultiplexing was carried out using Illumina’s bcl2dastq tool. Read alignment against hg38 human reference genome build and quality metrics were automatically generated using BWA-MEM/Picard pipeline and reviewed through the Yale Centre for Genome Analysis Tracking System software. Alignment and variant calling in the WGS data followed GATK v4 best practice guidelines. Variant annotation, filtration and prioritization was carried out using Exomiser v12.1.0 [27].
Results
Gene coverage descriptive statistics using smMIPs
A total of 903 smMIPs capturing the exons and intron–exon boundaries of 23 genes associated with 29 common LSDs were successfully designed (Additional file 2). We assessed the preliminary analytical performance of the assay in test samples (n = 3) that were previously genetically diagnosed for a given LSD. We analyzed the intra- and inter-sample uniformity of sequence coverage across all 23 genes. We sequenced 99.2% of the targeted region of approximately 53.7 kb with a mean (median) coverage after duplicate read removal of 536x (209x) during the first sequencing run. 147 smMIPs gave less than 30 reads during the first run. Upon rebalancing the probe pool, whereby tenfold increase in concentration of smMIPs having less than 30 reads in the first run was carried out, the mean (median) coverage was 442x (361x) with only 54 probes having less than 30 reads (Additional file 3). Approximately 0.8% of the targeted region which consisted of 5 genes- GALNS (2%), GBA (8.5%), IDS (2%), IDUA (4%), and SMPD1 (2.5%), was not covered by any smMIP probes. This was because of either sequence similarity with a known pseudogene or presence of low-complexity sequence region. Specifically, exons 5 and 11 of the GBA gene have a high sequence similarity with the pseudogene GBAP1, which were analysed by Sanger sequencing in samples with clinical suspicion of Gaucher disease. Of the 350 DNA samples tested, all were successfully processed at the first sequencing effort leading to a high-quality sequencing result for 100% of the samples tested in the validation and diagnostic yield cohort.
Assay validation
First, we evaluated the accuracy of our assay in detecting SNVs and CNVs by screening a cohort of 50 patients with known genetic aberrations, who were diagnosed using biochemical assay followed by conventional Sanger sequencing of a given LSD gene (Table 2). smMIP assay data analysis was done in a blinded fashion, and only after the result of the smMIP assay was interpreted, we compared the results to those of the conventional methods.
Overall, our smMIP based assay gave concordant results with conventional methods in 98% of the cases (n = 49/50; Table 2). All of the SNVs in the previously diagnosed samples, except for one sample, were accurately identified by our assay. The single discordant case was “sample 5” where the variant c.107 T > G was not detected in the GALNS gene. The smMIP assay failed to detect this variant as the variant was present in a low-complexity region of the gene for which smMIP probes were not designed by the MIPgen tool. Of interest, the smMIP assay detected a multi-exon deletion in the IDS gene in “sample 40” which was previously not detected by conventional methods. The smMIP assay result was validated by end-point PCR. Additionally, in “sample 36”, the smMIP assay detected compound heterozygous variants c.955+3G>A and c.3182T>C in the NPC1 gene. Interestingly, the prior variant was undetected by the conventional method, hence, the smMIP assay could provide complete genetic diagnosis for this sample.
We also assessed the strength of our assay in detecting CNVs. 10% of the samples (n = 5/50) had prior diagnosis of deletions in the HEXA gene (n = 4/5) and GALC gene (n = 1/5). Our smMIP-based assay correctly identified two homozygous HEXA deletions, two heterozygous HEXA deletions, and one heterozygous GALC deletion, which was consistent with the previous diagnosis (Table 2). Overall, our assay detected 97.9% SNVs and 100% CNVs in the validation cohort samples.
Diagnostic yield in enzymatically confirmed cases of LSD
We assembled a cohort of 187 patients that had received diagnosis for one of the 29 common LSDs through biochemical tests only. The classification of all samples based on the LSD sub-types were as following: mucopolysaccharidoses (n = 60/187; 32.2%), sphingolipidoses (n = 89/187; 47.8%), glycogen storage disease (n = 7/187; 4%), neuronal ceroid lipofuscinoses (n = 14/187; 8%), and post-translational modification defects (n = 17/187; 8.6%). For all these patients, clinical data and biochemical enzyme test results were collected and are presented in Additional file 4 and was used to carry out data analysis of the smMIP-assay.
Our analysis led to a confirmed genetic diagnosis in 156 of the 187 enzymatically confirmed cases (83.4%) with the presence of pathogenic or likely pathogenic variants in the targeted regions. For SNVs, the smMIP assay could detect all types of variants in the targeted region, with missense variants being the predominant variant type- missense (63%), nonsense (9%), splice site (11%), and small insertions/deletions (17%). Of 156 cases with a confirmed genetic diagnosis, CNVs spanning from single to multiple exons were observed in 8 cases (5%). This included 4 multi-exon deletions: 2 in the GALNS gene, one in the IDUA gene, and an exon 1–5 deletion in the HEXB gene. Single exon deletions were detected in the GALC, HEXB, IDS and PPT1 genes, one case of each (Fig. 1). Overall, we found CNVs in 5% of the total molecularly diagnosed cases in the present cohort. Of the 25 patients without molecular genetic diagnosis, we detected heterozygous pathogenic variant in the targeted regions of GLB1 and GNPTAB genes in 2 patients. These patients were enzymatically diagnosed as GM1 gangliosidosis and Mucolipidosis II/III, respectively. However, a second heterozygous variant in trans was not detected by the smMIP-assay, possibly suggestive of the variant present in the deep intronic region or presence of a complex structural variant; both of which are not detectable by the present assay. Figure 2 shows overview of the diagnostic yield achieved by the smMIP-based assay across 19 out of the 29 LSDs studied. We observed a 100% diagnostic yield in cases with the following LSDs: MPS IIIB (n = 5/5), Fabry disease (n = 2/2), and Niemann-pick type-C (n = 2/2) and 94% diagnostic yield in GM1 gangliosidosis cases (n = 17/18).

Automated visualizations of copy number variants from the DECoN tool. The top plot shows log normalised coverage of sample of interest (blue) relative to the reference samples (grey). Bottom plot shows ratio of observed to expected coverage. Relevant genes shown in between the plots in red. A Sample showing a homozygous exon 5 deleted in the HEXB gene. B Sample showing a homozygous exon 1–5 deleted in the HEXB gene. C Sample showing a homozygous exon 10–14 deleted in the GALNS gene. D Sample showing a homozygous exon 3–14 deleted in the IDUA gene. Deleted exons are highlighted in red

Diagnostic yield observed by smMIP-based NGS assay in the cohort with an enzyme diagnosis for a particular LSD
The smMIP-based assay identified causative variant(s) in 8 out of 9 cases each with either MPS I, MPS IIIA, MPS VI and Tay-Sachs disease thereby giving a diagnostic yield of 89% in these disease types. Additionally, the assay had a diagnostic yield of 86.6%, 73%, 71.4% and 88% for MPS IVA, Gaucher disease, Pompe disease and Mucolipidosis II/III disease, respectively. In 15 cases where no causative variant(s) were identified by the smMIP-based assay, whole exome/genome sequencing was carried out in order to assess if the variant(s) were present in deep intronic regions or were complex structural variants which would be missed by the smMIP based assay. On analysis of 15 cases, only a single case could be resolved whereby the case was enzymatically diagnosed to have Tay-Sachs disease and a single heterozygous variant c.902T>G in the HEXA gene was previously detected with the smMIP based assay (Table 3). We detected a deep intronic heterozygous variant c.413-358del in intron 3 of the HEXA gene, which was in trans with the aforementioned variant in this case. However, due to the lack of in vivo functional evidence for the intronic variant, the variant was classified as a variant of uncertain significance.
Poor diagnostic yield with the smMIP based assay was observed for MPS II cases whereby the yield was only 30.8% (n = 4/13). As the exon 3 of the IDS gene is not targeted by the smMIP-based assay due to its high sequence similarity with the pseudogene IDSP1, the remaining 8 cases were subjected to Sanger sequencing for exon 3. This led to diagnosis in 4 cases, leading to an overall genetic diagnosis in 62% of the MPS II cases (n = 8/13; Additional file 4). For the remaining 4 cases, no causative variants could be identified with whole exome sequencing (Table 3).
Of note, we observed 3 cases (sample ID: LSD1, LSD185 and LSD91) where there was discordance between the genetic diagnosis from smMIP based assay and biochemical assay. In 2 cases where the biochemical diagnosis of Gaucher disease was made due to the low levels of beta-glucosidase enzyme in leukocytes, the smMIP based assay detected pathogenic variant in the NPC1 and NPC2 gene in either case. In the third case with diagnosis of MPS I based on the biochemical assay, the smMIP-based assay detected no causative variants in the IDUA gene. In fact, a homozygous variant c.3503_3504del (p.Leu1168Glnfs*5) in the GNPTAB gene was observed which led to the genetic diagnosis of Mucolipidosis II/III. Thus, using the smMIP-based assay in all three cases, we could rectify the previous misdiagnosis of Gaucher disease as a case of Niemann Pick type C and MPS I as a case of Mucolipidosis II/III.
Diagnostic yield in cases with a clinical suspicion of LSD
We assessed 113 cases clinically suspected with one of the 29 LSDs using the smMIP-based assay (Additional file 5). We stratified these patients into “high-index” and “low-index” clinical suspicion groups based on the likelihood rank for one of the 29 LSDs using the phenotype scoring tool- GDDP (https://gddp.research.cchmc.org/). Patients where the rank was 1 to 15 were stratified to the “high-index” group (n = 73) and those with the rank > 15 were stratified to the “low-index” group (n = 40). A significantly higher diagnostic yield was observed in the high-index group (n = 54/73; 73.9%) compared to the low-index group (n = 1/41; 2.4%) using the smMIP-based assay (Additional file 5). The majority of the cases diagnosed in this entire cohort of patients belonged to mucopolysaccharidosis (n = 30); chiefly- MPS IIIB (n = 10), MPS II (n = 7), MPS IVA (n = 7), 4 cases of MPS IIIA, 3 cases of MPS I, and 2 cases of MPS VI. Using this assay, we identified causative variants in the GBA gene and SMPD1 gene in six patients and four patients respectively (Fig. 3).

Disease wise distribution of patients diagnosed by smMIP-based NGS assay in the clinical suspicion cohort
Importantly, with this assay we could provide a genetic diagnosis for Niemann pick type C and neuronal ceroid lipofuscinosis-6 for which currently, no biochemical tests are available. Overall, we identified 5 patients with Niemann pick type C, despite these patients being clinically suspected with Gaucher disease or Niemann-pick disease A/B.
Lastly, 17 cases in the high-index group where no causative variant(s) were identified using the smMIP based assay were subjected to WES (Table 4). Of note, 3 out of 17 cases were diagnosed with rare LSDs-Sialidosis type I/type II (OMIM#256550), Wolman disease (OMIM#620151) and GM2 gangliosidosis AB variant (OMIM#272750)-which are not covered by the smMIP based assay due to their low prevalence in the Indian population [19]. In further 7 cases, diseases not associated with LSDs were identified- progressive pseudorheumatoid dysplasia (OMIM#208230), intellectual developmental disorder 23 (OMIM#615761), hypermanganesemia with dystonia-1 (OMIM#613280), Neurodevelopmental disorder with or without hypotonia, seizures, and cerebellar atrophy (OMIM#616917), Beck-Fahrner syndrome (OMIM#618798) and microcephaly, short stature, and limb abnormalities (OMIM#617604) (Table 4).
Performance of smMIP based assay in DBS samples
Three patients whose blood sample was available for DBS and previously received genetic diagnosis using smMIP based assay was analysed for a comparative sequencing quality performance. Compared to 100 ng of input DNA extracted from blood, 20 ng of DNA was used for targeted capture and subsequent sequencing using smMIP based assay. Whilst no difference was observed in the percentage of mapped reads inside target region (96.9% for DNA from blood versus 97.4% for DNA from DBS), a higher proportion of duplicate reads based on UMB were detected (38% for DNA from blood versus 45% for DNA from DBS) (Additional file 6). With 80% reduction in the input DNA quantity, we observed a 38% drop in average sequence coverage across samples sequenced from DBS (160x) versus whole blood (258x) (Additional file 7). Despite the loss of coverage, no significant loss in variant calling accuracy across the samples was observed and genetic diagnosis could be made with 100% concordance.
Discussion
Diagnosis for LSDs is challenging due to several factors like phenotypic variability, the presence of overlapping clinical features across some LSDs, genetic heterogeneity and the difficulties associated with biochemical tests [2]. Recently, several studies have highlighted the incorporation of targeted NGS technologies as a potential diagnostic tool for LSDs [10, 11]. The essential advantage of using this approach includes unbiased interrogation of several genes at a time, thus enabling us to monitor a broader spectrum of diseases in a single test. This is especially beneficial in patients where the symptoms are not specific for a particular LSD and for LSDs where biochemical tests are not available. Fernandez-Marmiesse et al. for the first time demonstrated the use of a targeted sequencing assay to test 57 LSDs associated genes using in-solution capture as the enrichment method [10]. In the present study, we developed and applied a novel smMIP-based sequencing assay for the diagnosis of 29 common LSDs in India. We successfully demonstrated its ability to detect genetic abnormalities including both SNVs and CNVs by subjecting patient samples with previously identified genetic etiology and high clinical likelihood for one of the 29 common LSDs to the smMIP based assay study.
Despite a high proportion of targeted regions covered by the assay (99.2% of 53.7 kb), poor coverage was observed for genes (particularly IDS, IDUA and GBA) with low sequencing complexity or high sequence similarity with their pseudogene [33, 34]. This un-equivalency in target capture and sequencing of these genes is in congruence with observations made previously by Zanetti et al. [34]. For example, we observed poor diagnostic yield in clinically suspected MPS II cases. As exon 3 of the IDS gene is known to have a high sequence similarity with the pseudogene (IDSP1), no smMIP probes could be designed to capture this region with high specificity. Indeed, 4 patients received a genetic diagnosis of MPS II after Sanger sequencing was used to sequence exon 3 of the IDS gene in patients where the smMIP based assay didn’t identify causative variant. Like most NGS based assays (WES/WGS), one particular limitation of this assay is its inability to detect complex structural rearrangements. For example, smMIP-based assay cannot resolve and detect IDSP1-mediated IDS gene inversions or the RecNciI allele in the GBA gene, which is formed due to a non-homologous cross over between GBA and GBAP1 genes. Indeed, recent guidelines for genetic testing of these genes recommend Sanger sequencing of poorly covered regions or regions with high sequence similarity with pseudogene [13]. Additionally, orthogonal methods such as PCR–RFLP HinfI assay are suggested to be used in the detection of IDS/IDSP1 gene inversions in genetically undiagnosed MPS II patients as mentioned previously [35].
Importantly, the smMIP-based assay has high sensitivity and specificity for detection of both SNVs and CNVs due to the availability of UMBs in the backbone of the smMIP probes. This is reflected in the 98% and 100% concordance in SNV and CNV calling in the validation cohort. Furthermore, assessment of the assay’s diagnostic yield in a cohort of 300 patient samples ranged from 2.4 to 83%. This large variance in diagnostic yield is due to the heterogeneity of the patient cohort which consisted of 187 patient samples which had previously been diagnosed LSD using biochemical assay, 72 patient samples with a high clinical likelihood of LSD and 41 patient samples with a low clinical likelihood of LSD. Indeed, patients with prior biochemical assay based diagnosis or a high clinical suspicion of LSD showed a remarkable diagnostic yield of 83% and 74%, respectively, in comparison of the low clinical suspicion group, which showed yield of only 2.4%. This likely signifies and further emphasizes requirement of a deep clinical phenotyping before the used of NGS based assays in order to receive high diagnostic yields. Of note, observed diagnostic yields in both biochemically confirmed cases and high clinical suspicion cases are significantly higher than the yield of 62% in biochemically confirmed cases by Zanetti et al. [34]. The observed yields are also higher than that the reported yield of 67% by Di Fruscio et al. using Lysoplex in a group of 48 NCL patients [9] and 30% yield obtained using WES on 14 patients with an LSD suspicion reported by Wang et al. 2017 [36]. The higher diagnostic yield achieved in the high-index cohort in the present study is likely because of the deep clinical characterization of the patients before referring them for NGS-based panel studies. Interestingly, the type of disease for which a gene panel is offered also influences its diagnostic yield. For instance, the yield for a hypertrophic cardiomyopathy panel is as high as 32% [37] but for a congenital glycosylation disorders gene panel, it is only 14.8% [38]. This suggests that the complexity of the disease nature in question and its clinical presentation dictate the diagnostic success of gene panels. LSDs, in general, may present with a more specific phenotype. This explains the variability in diagnostic yield reported by different NGS panel studies for LSDs. It ranges from 15% reported by Gheldhof et al. in a cohort of 150 cases to 40% reported by Fernandez-Marmiesse et al. 2014 in a group of 66 suspected LSD patients [10, 39]. In addition, the smMIP-based assay could detect multi-exon and single-exon deletions in eight cases (~ 5%) of the total diagnosed cases. Large deletions in ~ 3–5% of cases of MPS II, Krabbe, and Niemann-pick diseases have been observed previously in the literature [40,41,42]. This observation is further strengthened by the in-ability to further improve diagnostic yield for 29 targeted LSDs using WES or WGS in patients where the smMIP based assay didn’t detect a causative variant.
An interesting observation was made for 3 biochemically diagnosed cases where the smMIP based assay corrected previous misdiagnoses. In two cases, biochemical diagnosis suggested Gaucher's disease, however, the smMIP based assay identified a causative variant in the NPC1 gene. Previously, it has been known that Niemann Pick type C is a differential diagnosis for Gaucher disease and is associated with falsely low beta-glucosidase activity [43]. Likewise, for another case with a reduced activity of alpha-iduronidase enzyme activity, a diagnosis of MPS I was made. However, the smMIP-based assay detected a variant in the GNPTAB gene. Patients with a defect in the GNPTAB gene display reduced activity of multiple lysosomal enzymes as there is a defect in the enzyme GlcNAc-1-phosphotransferase [44]. This enzyme is critical for tagging mannose-6-phosphate (M6P) to lysosomal enzymes so that they can bind to the M6P receptors on the trans-Golgi network [44]. Hence, patients with Mucoplipidosis-II/III can easily be misdiagnosed as MPS cases. Thus, genetic testing following biochemical testing is critical in such cases and the above observations highlight the strength of the assay in providing a diagnosis in cases with clinical heterogeneity. Previously, using this assay, we could also identify MLD due to activator protein deficiency in an adult patient [45], which could have been missed by a biochemical assay as these patients show normal levels of the arylsulfatase-A enzyme activity (Table 1). Thus, the assay can aid in the diagnosis of diseases like Niemann-Pick type C1/C2, saposin A/B/C deficiency as well as neuronal ceroid lipofuscinosis type 6, for which there are no well-established biochemical diagnostic tests available (Table 1).
Lastly, comparative analysis of the sequencing quality between DNA extracted from whole blood and DBS sample suggests potential utility of the smMIP based assay for newborn screening programs for detection of common LSDs in a given population. However, unlike whole blood samples, further evaluation and optimisation of the assay parameters for DBS samples may be warranted before its utilization in a clinical setting.
The costs of our smMIP-based assay are relatively low compared to the currently employed diagnostic pathway consisting of biochemical testing for LSD diagnosis. Although smMIPs require a relatively high initial investment, the per-patient library preparation and sequencing cost is estimated to be as low as US$73 on the Illumina MiSeq platform with Micro v2 flowcell and 200 × average sequence depth. This equates to approximately US$3.2 per gene per sample tested. However, in order to draw a definitive conclusion, further evaluation of cost-effective analysis needs to performed by comparing it with the costs incurred using the existing diagnostic route as well as calculating the time taken to reach to a diagnosis. Nonetheless, with the combined ability to detect both SNVs and CNVs, ease of use, high diagnostic yield and low costs, the utility of smMIP-based assay for 29 common LSDs irrespective of the clinical phenotype, especially in low-middle income countries, may allow for a paradigm shift in the clinical diagnostic pathway. Due to these advantages, clinical implementation of smMIP based NGS assays have previously been carried out in somatic microsatellite instability testing in colorectal cancer [46] and germline BRCA gene testing for identification of patients with hereditary breast and ovarian cancer [47].
Conclusions
We describe a novel and cost-efficient assay for genetic diagnosis of 29 common LSDs. We have shown that the assay can detect both SNVs and CNVs, and can be applied on DNA extracted from whole blood and DBS samples. The assay has proved to a powerful addition to the current diagnostic assay repertoire, and both patients and doctors can benefit greatly from utilizing this technique, especially in resource-limited settings.
Abbreviations
- LSDs:
-
Lysosomal storage disorders
- smMIPs:
-
Single-molecule molecular inversion probes
- ERT:
-
Enzyme replacement therapy
- NGS:
-
Next generation sequencing
- MLPA:
-
Multiplex ligation probe dependent amplification
- CNV:
-
Copy number variations
- SNV:
-
Single nucleotide variants
- UMB:
-
Unique molecular barcode
- LMICs:
-
Low-middle income countries
- ACMG:
-
American College of Medical Genetics
- DBS:
-
Dried blood spot
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Kingma SDK, Bodamer OA, Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract Res Clin Endocrinol Metab. 2015;29(2):145–57.
Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primer. 2018;4(1):27.
Sheth J, Pancholi D, Mistri M, Nath P, Ankleshwaria C, Bhavsar R, et al. Biochemical and molecular characterization of adult patients with type I Gaucher disease and carrier frequency analysis of Leu444Pro - a common Gaucher disease mutation in India. BMC Med Genet. 2018;19(1):178.
Sheth J, Nair A, Bhavsar R, Godbole K, Datar C, Nampoothiri S, et al. Lysosomal storage disorders identified in adult population from India: experience of a tertiary genetic centre and review of literature. JIMD Rep. 2024. https://doi.org/10.1002/jmd2.12407.
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–86.
Sheth J, Nair A. Treatment for lysosomal storage disorders. Curr Pharm Des. 2020;26(40):5110–8.
Agarwal S, Lahiri K, Muranjan M, Solanki N. The face of lysosomal storage disorders in India: a need for early diagnosis. Indian J Pediatr. 2015;82(6):525–9.
Sheth J, Nair A, Jee B. Lysosomal storage disorders: from biology to the clinic with reference to India. Lancet Reg Health Southeast Asia. 2023;9:5.
Di Fruscio G, Schulz A, De Cegli R, Savarese M, Mutarelli M, Parenti G, et al. Lysoplex: an efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy. 2015;11(6):928–38.
Fernández-Marmiesse A, Morey M, Pineda M, Eiris J, Couce ML, Castro-Gago M, et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J Rare Dis. 2014;25(9):59.
La Cognata V, Cavallaro S. A comprehensive, targeted NGS approach to assessing molecular diagnosis of lysosomal storage diseases. Genes. 2021;12(11):1750.
Encarnação M, Coutinho MF, Silva L, Ribeiro D, Ouesleti S, Campos T, et al. Assessing lysosomal disorders in the NGS era: identification of novel rare variants. Int J Mol Sci. 2020;21(17):6355.
Zampieri S, Cattarossi S, Bembi B, Dardis A. GBA Analysis in next-generation era: pitfalls, challenges, and possible solutions. J Mol Diagn JMD. 2017;19(5):733–41.
Pogoda M, Hilke FJ, Lohmann E, Sturm M, Lenz F, Matthes J, et al. Single molecule molecular inversion probes for high throughput germline screenings in dystonia. Front Neurol. 2019;10:1332.
Waalkes A, Penewit K, Wood BL, Wu D, Salipante SJ. Ultrasensitive detection of acute myeloid leukemia minimal residual disease using single molecule molecular inversion probes. Haematologica. 2017;102(9):1549–57.
Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23(5):843–54.
Oud MS, Ramos L, O’Bryan MK, McLachlan RI, Okutman Ö, Viville S, et al. Validation and application of a novel integrated genetic screening method to a cohort of 1,112 men with idiopathic azoospermia or severe oligozoospermia. Hum Mutat. 2017;38(11):1592–605.
Sheth J, Patel P, Sheth F, Shah R. Lysosomal storage disorders. Indian Pediatr. 2004;41(3):260–5.
Sheth J, Mistri M, Sheth F, Shah R, Bavdekar A, Godbole K, et al. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014;12:51–63.
Verma PK, Ranganath P, Dalal AB, Phadke SR. Spectrum of Lysosomal storage disorders at a medical genetics center in northern India. Indian Pediatr. 2012;49(10):799–804.
Bean LJH, Funke B, Carlston CM, Gannon JL, Kantarci S, Krock BL, et al. Diagnostic gene sequencing panels: from design to report-a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med Off J Am Coll Med Genet. 2020;22(3):453–61.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Boyle EA, O’Roak BJ, Martin BK, Kumar A, Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014;30(18):2670–2.
O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–22.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2009;25(14):1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Smedley D, Jacobsen JOB, Jäger M, Köhler S, Holtgrewe M, Schubach M, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc. 2015;10(12):2004–15.
Fowler A. DECoN: a detection and visualization tool for exonic copy number variants. Methods Mol Biol Clifton NJ. 2022;2493:77–88.
Biesecker LG, Harrison SM, ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med Off J Am Coll Med Genet. 2018;20(12):1687–8.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. 2015;17(5):405–24.
Zhang J, Yao Y, He H, Shen J. Clinical interpretation of sequence variants. Curr Protoc Hum Genet. 2020;106(1): e98.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinform Oxf Engl. 2012;28(21):2747–54.
Málaga DR, Brusius-Facchin AC, Siebert M, Pasqualim G, Saraiva-Pereira ML, de Souza CFM, et al. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet Mol Biol. 2019;42(1 suppl 1):197–206.
Zanetti A, D’Avanzo F, Bertoldi L, Zampieri G, Feltrin E, De Pascale F, et al. Setup and validation of a targeted next-generation sequencing approach for the diagnosis of lysosomal storage disorders. J Mol Diagn JMD. 2020;22(4):488–502.
Alcántara-Ortigoza MA, García-de Teresa B, González-Del Angel A, Berumen J, Guardado-Estrada M, Fernández-Hernández L, et al. Wide allelic heterogeneity with predominance of large IDS gene complex rearrangements in a sample of Mexican patients with Hunter syndrome. Clin Genet. 2016;89(5):574–83.
Wang N, Zhang Y, Gedvilaite E, Loh JW, Lin T, Liu X, et al. Using whole-exome sequencing to investigate the genetic bases of lysosomal storage diseases of unknown etiology. Hum Mutat. 2017;38(11):1491–9.
Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med Off J Am Coll Med Genet. 2015;17(11):880–8.
Jones MA, Rhodenizer D, da Silva C, Huff IJ, Keong L, Bean LJH, et al. Molecular diagnostic testing for congenital disorders of glycosylation (CDG): detection rate for single gene testing and next generation sequencing panel testing. Mol Genet Metab. 2013;110(1–2):78–85.
Gheldof A, Seneca S, Stouffs K, Lissens W, Jansen A, Laeremans H, et al. Clinical implementation of gene panel testing for lysosomal storage diseases. Mol Genet Genomic Med. 2019;7(2): e00527.
Agrawal N, Verma G, Saxena D, Kabra M, Gupta N, Mandal K, et al. Genotype-phenotype spectrum of 130 unrelated Indian families with Mucopolysaccharidosis type II. Eur J Med Genet. 2022;65(3): 104447.
Irahara-Miyana K, Enokizono T, Ozono K, Sakai N. Exonic deletions in GALC are frequent in Japanese globoid-cell leukodystrophy patients. Hum Genome Var. 2018;5:28.
Hebbar M, Prasada LH, Bhowmik AD, Trujillano D, Shukla A, Chakraborti S, et al. Homozygous deletion of exons 2 and 3 of NPC2 associated with Niemann-Pick disease type C. Am J Med Genet A. 2016;170(9):2486–9.
Lo SM, McNamara J, Seashore MR, Mistry PK. Misdiagnosis of Niemann-Pick disease type C as Gaucher disease. J Inherit Metab Dis. 2010;33 Suppl 3(03):S429-433.
Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4(3):202–12.
Sheth J, Nair A, Bhavsar R, Shah H, Tayade N, Prabha CR, et al. Late infantile and adult-onset metachromatic leukodystrophy due to novel missense variants in the PSAP gene: case report from India. JIMD Rep. 2023;64(4):265–73.
Gallon R, Sheth H, Hayes C, Redford L, Alhilal G, O’Brien O, et al. Sequencing-based microsatellite instability testing using as few as six markers for high-throughput clinical diagnostics. Hum Mutat. 2020;41(1):332–41.
Neveling K, Mensenkamp AR, Derks R, Kwint M, Ouchene H, Steehouwer M, et al. BRCA testing by single-molecule molecular inversion probes. Clin Chem. 2017;63(2):503–12.
Acknowledgements
We are grateful to all the patient families for their kind co-operation and permission. The authors would like to thank Joris Veltman, Biosciences Institute, Newcastle University for scientific and technical support.
Funding
We sincerely acknowledge the research grant from the Department of Biotechnology, Government of India- (DBT BT/PR39587/MED/12/851/2020) and Gujarat State Biotechnology Mission, Department of Science and Technology (GSBTM-DST/JDR D/608/2020/459-461) for funding this work. The funders had no role in the study design, sample collection and data analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed experiments: HS, JS, AN, CJ and MJ. Patient recruitment and clinical analysis: MK, KNVR, AB, SK, SN, IP, AK, SS, SM, SJ, IS, SK, SB, RRD, AP, KG, HP, ZL, RS, AI, SB, RP, MM, AS, SM, NG, NT, AG, AS, AJ, DS, AD, FS and JS. Target capture and library preparation: AN, RB, SK and HS. Sequencing: HS, AN, CJ and MJ. Data analysis and interpretation: HS, AN and JS. Write first draft of the manuscript: AN, HS and JS. Made critical revisions and approved final version: JS, HS, CRP. All authors reviewed and approved the final manuscript. HS and JS accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the institutional ethics committee of FRIGE’s Institute of Human Genetics (Reg No- E/13237) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients before inclusion in the study.
Consent for publication
Not applicable.
Competing interests
Harsh Sheth, Aadhira Nair and Jayesh Sheth are named as inventors on the patent describing the use of smMIP based target capture and associated computational analyses for simultaneous detection of single nucleotide variants and copy number variants in germline DNA. The patent is held by FRIGE’s Institute of Human Genetics (Patent ID: TEMP/E-1/30548/2022-MUM, submitted in May 2022). All other authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. List of study sites and recruited patients.
Additional file 2
. Single molecule molecular inversion probe sequences of 903 probes along with their target enrichment site coordinates used in the smMIP based assay.
Additional file 3
. Overall distribution of reads across the 903 smMIP molecules before and after rebalancing the smMIP pool. Considering, total 903 probes, we expect 0.001 proportion of reads to be the optimum value by each probe. The navy blue denotes the optimum proportion of read value i.e 0.001. A lower cut-off value of 0.0001 and higher cut-off value of 0.01 was set as the optimum range to assess the efficiency of the probes.
Additional file 4
. Clinical data and biochemical assay results of 187 patients with prior biochemical diagnosis of LSD in the diagnostic yield cohort.
Additional file 5
. Clinical data and smMIP based assay result in 113 patients with a clinical suspicion of LSD in the diagnostic yield cohort.
Additional file 6
. Comparative data for Dried Blood Spot (DBS) extracted DNA and Manual blood extracted DNA used in smMIP assay.
Additional file 7
. Coverage data of 903 probes using Dried Blood Spot (DBS) extracted DNA and Manual blood extracted DNA in smMIP-NGS assay.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sheth, H., Nair, A., Bhavsar, R. et al. Development, validation and application of single molecule molecular inversion probe based novel integrated genetic screening method for 29 common lysosomal storage disorders in India. Hum Genomics 18, 46 (2024). https://doi.org/10.1186/s40246-024-00613-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-024-00613-9