
Abstract
Background
High-quality genomic datasets from under-representative populations are essential for population genetic analysis and medical relevance. Although the Tujia are the most populous ethnic minority in southwestern China, previous genetic studies have been fragmented and only partially reveal their genetic diversity landscape. The understanding of their fine-scale genetic structure and potentially differentiated biological adaptive features remains nascent.
Objectives
This study aims to explore the demographic history and genetic architecture related to the natural selection of the Tujia people, focusing on a meta-Tujia population from the central regions of the Yangtze River Basin.
Results
Population genetic analyses conducted on the meta-Tujia people indicate that they occupy an intermediate position in the East Asian North-South genetic cline. A close genetic affinity was identified between the Tujia people and neighboring Sinitic-speaking populations. Admixture models suggest that the Tujia can be modeled as a mixture of northern and southern ancestries. Estimates of f3/f4 statistics confirmed the presence of ancestral links to ancient Yellow River Basin millet farmers and the BaBanQinCen-related groups. Furthermore, population-specific natural selection signatures were explored, revealing highly differentiated functional variants between the Tujia and southern indigenous populations, including genes associated with hair morphology (e.g., EDAR) and skin pigmentation (e.g., SLC24A5). Additionally, both shared and unique selection signatures were identified among ethnically diverse but geographically adjacent populations, highlighting their extensive admixture and the biological adaptations introduced by this admixture.
Conclusions
The study unveils significant population movements and genetic admixture among the Tujia and other ethno-linguistically diverse East Asian groups, elucidating the differentiated adaptation processes across geographically diverse populations from the current genetic landscape.
Similar content being viewed by others


Introduction
East Asia is characterized by some of the highest levels of ethnic, linguistic, and cultural diversity in the world. The earliest evidence of ancient humans in the region dates back at least 1.6 million years, with anatomically modern humans having migrated out of Africa approximately 50,000 to 70,000 years ago [1, 2]. The region is home to diverse language families, including Altaic, Sino-Tibetan (ST) [Sinitic and Tibeto-Burman (TB)], Hmong-Mien (HM), Tai-Kadai (TK), Austronesian (AN), and Austroasiatic (AA). Conducting comprehensive population genetic research on underrepresented ethnolinguistic groups can offer valuable insights into their origins, migration patterns, admixture, and medical relevance [3,4,5,6]. Additionally, these studies can have critical translational applications in various fields [5]. Archaeological and anthropological research has revealed complex demographic dynamics within East Asia, which is strongly associated with geographical isolations and linguistic affiliations [3, 6, 7]. Remarkably, the ST language family, primarily comprising Sinitic and TB languages, represents the world’s second-largest language lineage and possesses a complex admixture landscape with different patterns from highland and lowland East Asia [6, 8]. Archaeological evidence supports the “Northern origin hypothesis” for the ST language family, linking its expansion to the Yangshao and Miaozigou cultures in Northern China’s Yellow River Basin (YRB). Recent genetic and linguistic studies have systematically reconstructed the formation and migration history of East Asians, corroborating the theory that the diffusion of agriculture coincided with language spread in the region [3, 6,7,8,9,10,11,12]. It was discovered that Neolithic farmers from the YRB are ancestors of TB-speaking populations [6]. Population genetic modeling has further demonstrated that the primary ancestry of Tibetan and Yi populations relates to these millet farmers, undergoing complex biological adaptations [6, 13]. Additionally, significant admixture events were identified as pivotal in shaping the genomic diversity of ethno-linguistically diverse lowland TB populations [6, 14]. This process involved the southward expansion of Neolithic farmers from the YRB through the Tibetan-Yi Corridor, merging with indigenous hunter-gatherer populations [6]. Throughout this period, TB-speaking populations also extensively interacted with TK/HM-speaking populations in southwestern China [15]. Both archaeological and molecular evidence suggests that Neolithic farmers played a crucial role in the formation of modern Tibetans, mainly through the core region of the Qinghai-Tibet Plateau [7, 16].
The Tujia people, a unique TB-speaking population, are widely distributed across central China. Previous genetic studies on this group have primarily utilized traditional forensic markers, such as short tandem repeats (STRs), low-density single nucleotide polymorphisms (SNPs), and variations in the Y chromosome and mitochondrial DNA (mtDNA), to investigate their genetic structure and relationships with other populations [17,18,19,20,21]. He et al. conducted a population genetic analysis of the Tujia people, focusing on shared alleles, which indicated a close genetic relationship with the Han Chinese [22]. However, previous research has often neglected the demographic history and adaptive landscape of these and other populations [14]. By analyzing shared alleles and haplotypes, a more refined understanding of the Tujia people’s genetic structure and adaptive history can be achieved, facilitating exploration of their interactions with neighboring, linguistically diverse populations such as Hmong-Mien, Turkic, Sinitic, and Austro-Asiatic speakers [6, 9, 10, 23]. Furthermore, this knowledge is vital for addressing questions about the origins, admixture, and complex migration and admixture histories in eastern Eurasia, as well as their implications for the health and disease susceptibility of the Tujia people.
Despite ongoing efforts to decipher the complex movement and admixture history of the TB-speaking Tujia people in East Asia, detailed knowledge of their fine-scale genetic structure and distinctive biological adaptive traits derived from high-density shared alleles and haplotypes remains elusive. While modern humans have evolved a variety of phenotypic characteristics unique to different ethnic groups, the genetic underpinnings of these adaptative physiological variations are not yet fully understood. The debate within the fields of physical anthropology and human evolutionary genetics centers on whether these traits have developed as adaptations to diverse local environments or through other evolutionary processes. Recent advances in the analysis of genomic data from various populations have emerged as a crucial method for addressing these pivotal questions [24,25,26,27,28]. In this study, we generated genome-wide data comprising nearly 700,000 SNPs from the central Chinese Tujia population and integrated them with existing public genetic datasets. This comprehensive dataset provides valuable insights into the Tujia’s geographically diverse population, facilitating further exploration of their fine-scale genetic structure, migration history, natural selection processes, and medical implications, as well as their genetic connections to surrounding ethnolinguistically distinct populations.

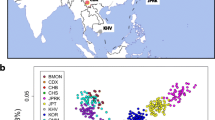
Geographical position and population structure of four Tujia populations. a The geographical locations of newly collected samples alongside reference populations are depicted (Table S1). b-d Principal component analysis (PCA) of the genetic patterns of modern and ancient East Asians was conducted using the merged Human Origins (HO) dataset. Ancient individuals were projected onto the PCA plot, while modern populations were color-coded according to their language family categories. Detailed PCA patterns with complete population labels can be found in Fig. S1. YRB: Yellow River basin; WLR: West Liao River basin; AR: Amur River region. e Results from the model-based ADMIXTURE clustering analysis at K = 8 are shown, with further details on the admixture models provided in Fig. S2
Results
Genetic clustering and population structure of the Tujia people
Approximately 700,000 genome-wide SNPs were genotyped in 18 Tujia individuals from Hubei Province, and these data were integrated with publicly available modern and ancient DNA datasets, including the Human Origins (HO), the 1240 K dataset, the Human Genomic Diversity Project (HGDP), the Oceania genomic resource, and our recently published East Asian dataset [6, 10, 12, 29]. Individuals were categorized based on their language family affiliations, encompassing modern populations from TB-speaking (n = 618), Sinitic-speaking (n = 687), TK-speaking (n = 286), HM-speaking (n = 130), AN-speaking (n = 120), AA-speaking (n = 236), and Altaic-speaking (n = 135) groups, while ancient individuals were classified by their geographical origins. Principal Component Analysis (PCA) was conducted to explore the fundamental patterns of genetic admixture and relationships between the Tujia people and other modern and ancient East Asian populations. The PCA utilized 123 modern populations to construct principal components, with 107 ancient populations projected onto the plot. PC1 elucidated the pattern of genetic differentiation between northern regions, represented by Altaic- and TB-speaking populations, and southern regions, represented by HM-, TK-, AN-, and AA-speaking populations (Fig. 1b). PC3 specifically distinguished HM-speaking populations from other East Asian groups (Fig. 1c). The results positioned Wendou Tujia (WDT) and three other Tujia populations at an intermediate position between northern East Asian (NEA) and southern East Asian (SEA) clines, partially overlapping with Sinitic-, TB-, and HM-speaking populations. Notably, a closer genetic affinity was identified between the Tujia groups and the YRB farmers compared to other ancient populations (Fig. 1d).
Additional PCA was conducted to assess the genetic affinity between the WDT population and 62 modern reference populations. Three genetic clusters were identified: a northern cluster, which includes TB-speaking populations and a northern Han population; a southern cluster composed of HM-speaking populations; and an intermediate cluster that encompasses AA-, AN-, and Sinitic-speaking populations, along with the four Tujia groups (Fig. S3a). The Shaanxi Han was positioned at the southernmost end of the northern cline, exhibiting a close relationship with the coastal Fujian Han people. The WDT and the three other published Tujia populations formed a coherent cluster consistent with their linguistic affiliations and partially overlapped with other previously published Sinitic-speaking populations. Additionally, PCA was performed to explore the patterns of relatedness among geographically diverse TB-speaking populations (Fig. S3b). It was observed that the WDT, along with other Tujia populations from three geographically diverse regions, formed a cluster positioned between the high-altitude and low-altitude TB clusters, suggesting nuanced regional genetic integrations within the TB-speaking groups.
To explore the ancestry composition and genetic patterns of the Tujia populations, we conducted a model-based ADMIXTURE analysis using data from 123 modern and 107 ancient populations in East Asia (Fig. 1e). Cross-validation was used to determine the optimal number of clusters and the analysis indicated that the admixture model with eight ancestral sources (K = 8) provided the best fit. The results demonstrated that the four geographically diverse Tujia populations exhibited similar ancestry patterns. A significant component, enriched in ancient YRB farmers (light blue), was also found to be prevalent in the southern Han Chinese population. The other ancestral components, associated with inland Gaohuahua and coastal populations such as Tanshishan, Xitoucun, and Hanben, were present in relatively smaller proportions compared to the components of ancient YRB farmers.
To elucidate the fine-scale ancestral composition and genetic affinity of the Tujia populations, we conducted a model-based ADMIXTURE analysis among 85 modern populations in East Asia (Fig. S3c). The “optimal” number of clusters, determined via cross-validation, was five (K = 5): one cluster was predominantly found in Sinitic-speaking populations (pink), another in TK-speaking populations (yellow), a third in TB-speaking populations (blue), with the remaining two clusters prevalent among HM-speaking populations (orange) and Bai/Manchu populations in Southwest China (green). The results indicated that the genetic makeup of the WDT population is closely aligned with that of TB-speaking populations in South China.

Estimates of outgroup f3 and admixture f3 statistics based on shared alleles for WDT. a The outgroup f3 statistics were calculated using the HO_Illumina dataset formatted as f3(Modern East Asian, WDT; Mbuti). b Admixture signals for WDT were assessed via admixture-f3(Modern East Asian 1, Modern East Asian 2; WDT), highlighting potential genetic contributions from two distinct East Asian populations. c Outgroup-f3 statistics were derived from the 1240K_Illumina dataset, formatted as f3(Ancient East Asian, WDT; Mbuti), to explore genetic affiliations with ancient populations. d Admixture-f3 analysis for WDT (Ancient East Asian 1, Ancient East Asian 2; WDT) identified significant admixture events. Admixture-f3 values exhibiting Z scores less than − 3 suggest that WDT was an admixture of two parental populations, designated as source 1 and source 2. Z scores less than − 3 are denoted by “*”, indicating varying degrees of admixture significance
Population relationships and genetic composition of Tujia
The genetic relationships and composition of the Tujia population were analyzed using outgroup f3 statistics, calculated as f3(source 1, WDT; Mbuti) to assess allele sharing between WDT and various modern and ancient reference populations. It was observed that the WDT population exhibited greater genetic affinity with Sinitic-speaking Han and Tanka populations, ST-speaking HGDP Tujia population, and HM-speaking Miao population (Fig. 2a). Among ancient reference populations, more alleles were shared with YRB farmers than with ancient southern East Asians (Fig. 2c). Additionally, the potential ancestral sources of Tujia were investigated using admixture-f3 statistics, represented as f3(source 1, source 2; WDT), where source 1 and source 2 included different modern and ancient East Asian groups. Significant admixture signals (Z < − 3) were identified when HM-speaking populations and Shaanxi Han were used as the SEA and NEA sources, respectively (Fig. 2b). Similarly, when ancient groups were considered, significant admixture signals (Z < − 3) were found with Hanben/BaBanQinCen as the ASEA source and YRB farmers/Amer River Basin/Ancient_Nepal populations (e.g., Kyang, Mebrak, and Samdzong) as the ANEA source (Fig. 2d). The results from admixture f3 statistics robustly indicated significant north-south admixture in WDT, underscoring complex historical interactions between these groups.
To elucidate the genetic differentiation between the WDT population and other groups, we computed pairwise fixation index (Fst) genetic distances among 85 East Asian populations (Fig. S4). It was found that WDT was genetically closer to the Sinitic/HM-speaking populations, demonstrating a greater genetic affinity for these groups. Additionally, pairwise Fst genetic distances were calculated among 12 TB-speaking populations, which included three previously published Tujia populations (Fig. 3c). WDT and the other three Tujia populations formed a distinct cluster compared to the remaining TB-speaking groups. To infer population split events and the phylogenetic relationships involving WDT and other groups, a phylogenetic tree was constructed among 48 East Asian populations using TreeMix analysis (Fig. 3a and Fig. S5). The analysis revealed that WDT and southern Sinitic populations occupied an intermediate position on the tree, situated between the northern branch, which included Altaic- and some highland TB-speaking populations, and the southern branch, which comprised southern HM-, TK-, AN-, and AA-speaking populations from southern China. However, with the increasing number of gene flow events, we identified no evidence of gene flow events into or out of the Tujia population.
Admixture scenarios and gene flows of the Tujia people
Admixture scenarios and gene flows of the Tujia people were investigated through the application of f4 statistics, assessing the genetic homogeneity and heterogeneity among the studied Tujia group and three other published groups, using symmetrical-f4 in the form of f4(geographically different Tujia, WDT; reference populations, Mbuti). No significant negative f4 values were observed in the analysis of f4(JKT/QJT/YHT, WDT; modern/ancient East Asians, Mbuti), indicating that WDT and the other three geographically distinct Tujia groups exhibit genetic homogeneity relative to other East Asians (Table S2a-b). Conversely, significantly positive f4 values in f4(JKT, WDT/QJT/YHT; HM-speaking populations, Mbuti) were identified, suggesting that HM-speaking populations share more alleles with JKT than with the other Tujia groups (Table S2c). These findings underscore a relative genetic homogeneity among the four geographically diverse Tujia populations.
To further investigate the gene flow of the Tujia with other populations, affinity-f4 statistics were calculated in the form of f4(reference population 1, reference population 2; WDT, Mbuti). It was observed that WDT shared more alleles with low-altitude ST-speaking populations than with high-altitude ST-speaking populations, as indicated by significant negative f4 values in f4(high-altitude ST-speaking populations, East Asians; WDT, Mbuti) (Table S3a). This analysis was similarly applied to three other Tujia groups, yielding analogous results. In the context of HM-related ancestry among Miao, She, and Yao populations, significantly positive f4 values were noted in f4(HM-speaking populations, East Asians; WDT, Mbuti), suggesting greater allele sharing between HM-speaking populations and WDT compared to ST/AA-speaking populations (Table S3b). Among AA-speaking populations, significant negative f4 values in f4(Blang/Wa, East Asians; WDT, Mbuti) suggested that WDT had more ancestry from Sinitic/HM-speaking populations than from AA-speaking populations (Table S3c). Conversely, positive f4 values were recorded in f4(Gaoshan, East Asians; WDT, Mbuti), indicating a closer genetic affinity between WDT and AN-speaking populations than with high-altitude ST-speaking populations (Table S3d). Analysis involving Mulam, Maonan, and Dong as representatives of TK-related ancestry showed significantly positive f4 values in f4(TK-speaking populations, East Asians; WDT, Mbuti), revealing that WDT shared more alleles with geographically proximate TK-speaking populations than with other geographically distant TK/AN/Mongolic-speaking populations (Table S3e). Lastly, to assess the genetic affinity between the Tujia population and geographically close TK/HM-speaking populations, f4 tests were conducted in the form of f4(HM-speaking populations, TK-speaking populations; WDT, Mbuti), resulting in significantly positive f4 values, which confirmed greater allele sharing between WDT and HM-speaking populations compared to TK-speaking populations (Table S3f).
The genetic continuity and admixture between the WDT and ancient populations were further investigated using f4 tests. The analysis indicated that WDT individuals exhibited a greater genetic affinity with more recent northern and southern ancient populations than with more distant ancients, as evidenced by significant negative f4 values in f4(Baojianshan/Longlin, ancient East Asians; WDT, Mbuti) (Table S4a). When ancient samples from spatiotemporally distinct YRB farmers were used as northern reference sources, predominantly negative f4 values were observed (ancient East Asians, WDT; ANEAs, Mbuti), suggesting a genetic association between WDT and ancient YRB farmer-related ancestry (Table S4b). Comparative analysis with ancient DNA further demonstrated that WDT harbored more Southeast Asian (ASEA) ancestry when these YRB farmers were used as the northern reference, as shown by significant negative f4 values (ANEAs, WDT; ancient East Asians, Mbuti). This supports the conclusion that the Tujia people derive ancestry from both northern and southern East Asia (Table S4c).

Population relationships and genetic composition of Tujia people. a The phylogenetic relationship among East Asian populations shows a close genetic affinity between the Tujia and both Sinitic-speaking groups. Populations are categorized by linguistic affiliation. Wa population was set as the outgroup. b Pairwise qpWave analysis reveals both genetic heterogeneity and homogeneity among modern Chinese populations. P-values from rank0 tests exceeding 0.05 denote genetic homogeneity between two reference populations, indicated by “++”; p-values above 0.01 are marked with “+”. c A heatmap displays the pairwise Fst genetic distances among 12 geographically diverse TB-speaking populations. d Estimates of mixture proportions derived from qpAdm are presented. Two-way admixture models suggest contributions from both North and South Asians to the genetic makeup of the Tujia people, with the white error bar representing the standard error
In light of the similar genetic patterns observed among the geographically different Tujia populations, a qpWave test was conducted on paired populations to confirm their homogeneity (Fig. 3b). Further pairwise qpWave analysis among 72 populations was performed to investigate the genetic relationships between the Tujia people and other modern ethnolinguistic groups. This analysis revealed strong genetic homogeneity between the geographically diverse Tujia and the Sinitic-speaking populations from South China. Additionally, genetic homogeneity was observed among populations that are both geographically and linguistically proximate, particularly between the Sinitic- and HM-speaking groups. This suggests a possible recent common origin or extensive population admixture among Han Chinese and Tujia people. An alternative model posits cultural diffusion, whereby the culture and traditional practices of the ancient Tujia were assimilated by the Han Chinese, leading to the formation of the modern, culturally defined Tujia people.
The qpAdm tool was used to analyze the admixture proportions using predefined modern northern and southern ancestral East Asian populations (Fig. 3d and Table S5). Two-way admixture models were fitted using Oroqen, Hezhen, and northern Han as modern northern ancestral sources and Yao and Li as southern ancestral sources. When northern Han was utilized as the northern source, it was observed that contemporary northern ancestral sources accounted for a larger proportion of the ancestry, ranging from 54.5 to 71.6%. Conversely, when Altaic speakers were used as the northern source, the SEA populations (Li) contributed more significantly, ranging from 65.9 to 72%, to WDT in the two-way admixture model. This divergence in the proportions of individuals with NEA ancestry may be attributed to the early divergence between Sino-Tibetan (northern Han) and Altaic speakers or the close relationship between the northern Han and the reliable ancestral northern source.
The analysis of identity by descent (IBD) fragments across various ranges indicated that four Tujia individuals demonstrated high genetic relatedness, as evidenced by a substantial number of shared IBD categories over the past 1,500 years. The consistency in the number and length of these shared IBD fragments aligns with patterns observed across different length ranges (Fig. 4a). A plausible population bottleneck in the ancient Tujia population, estimated to have occurred between 15 and 20 generations ago, is suggested by the estimated effective population sizes (Fig. 4b-c). Archaeological records reveal that the Yangtze River basin was frequently subjected to floods and wars during the Ming Dynasty. Notably, in the late Ming Dynasty, high population density and fragile societal conditions contributed to widespread and recurrent fatalities, predominantly due to the synergistic effects of climate change and various social factors (https://www.zjujournals.com/sci/CN/Y2022/V49/I1/85).

Estimation of identity by descent (IBD) and effective population size among geographically diverse TB-speaking populations. a The IBD sharing model is used to evaluate the average IBD segment length within ranges of 1–5 cM and over 5 cM, as well as the total count and length of IBD segments among 12 geographically diverse TB-speaking populations in the HO dataset. b Estimates of effective population sizes for six geographically diverse TB-speaking populations across the past 150 generations are presented. c The effective population sizes for four Tujia individuals are assessed over the past 150 generations, with each generation defined as spanning 29 years
Natural selection signatures among the Tujia people
Signatures of natural selection under different environments among Chinese populations have been elucidated through evolutionary genetic studies, revealing numerous biologically adaptive genes and pathways linked to environmental adaptation, pathogen exposure, and dietary changes across ethnolinguistically diverse groups [30]. Due to the significant genetic homogeneity among Tujia individuals compared with other ethnolinguistically different Chinese populations, we combined the four Tujia subgroups into a single meta-Tujia population for the population branch statistic (PBS) analysis. To identify high-confidence candidate gene sets under natural selection over long evolutionary timescales, we examined three distinct PBS models with different ingroups, and we employed a random sampling method to ascertain the significance of the leading candidate loci under selection [31, 32]. Using the top 0.1% of PBS values (statistically supporting deviations between the adaptive candidates and the neutral loci) and requiring the statistically supporting adaptative mutation site to be present at least twice as selection criteria, we identified 460 candidate SNPs associated with 36 genes, indicating strong selection signatures based on PBS(Tujia−Li−CEU) (Fig. 5a and Table S6a). The most pronounced selection signature was found in the Endomucin (EMCN) gene, located on chromosome 4. The EMCN gene encodes endomucin, a glycoprotein primarily expressed in endothelial cells, playing a critical role in maintaining vascular integrity and function. Endomucin exhibits anti-adhesive properties that help prevent thrombosis and is essential for angiogenesis and the formation of new blood vessels. Its extensive glycosylation influences endothelial cell behavior and interactions with other proteins and cellular components. Additionally, genes such as ZNF638, ZMYM4, PSMB2, and BST1 were identified, highlighting allele frequency differences between the Tujia and Li populations and underscoring the gradual changes in derived allele frequencies among geographically diverse Chinese populations.
PBS models with different ingroups have different resolutions for identified adaptive candidates in different divergence time scales. Using a model with inland TK-speaking Shui_Guizhou individuals as the ingroup reference and European individuals as the outgroup, we identified 460 candidate SNPs associated with 53 genes over the 99.9th percentile in the PBS values of the PBS(Tujia−Shui−CEU) model (Fig. S6a and Table S6b). Notably, we observed a strong selection signal in the RAN binding protein 3 like (RANBP3L) gene on chromosome 5. This gene encodes a protein involved in nucleocytoplasmic transport, functioning as an export receptor for the nuclear export of proteins and RNA. RANBP3L is essential for regulating nuclear import and export, thereby influencing various cellular processes such as cell cycle progression and gene expression. Additionally, RANBP3L modulates signaling pathways and may play a role in cancer development through its impact on cellular transport mechanisms.
Subsequently, we employed a geographically distinct inland TK population as an ingroup to compare our candidates in the PBS model. We used the Zhuang_Guangxi population as the ingroup reference and the European population as the outgroup reference to explore associated biological adaptive signatures. Filtering the PBS values over the 99.9th percentile distribution of PBS(Tujia−Zhuang−CEU) identified 460 candidate SNPs associated with 32 selected genes (Fig. S6b and Table S6c). We additionally identified a significant selection signal on chromosome 2 at the Ectodysplasin A receptor (EDAR) gene, which is crucial for the development and function of ectodermal tissues such as skin, hair, teeth, and sweat glands. Notably, the SNP rs3827760 within the EDAR gene, showing strong positive selection signatures in East Asians, marks pronounced genetic differentiation among populations. This SNP’s selection suggests it has been favored by natural selection for its effects on hair morphology, highlighting its adaptive importance in East Asian populations [30, 33,34,35]. Additionally, we identified a significant finding in the Solute Carrier Family 24 Member 5 (SLC24A5) gene on chromosome 15. This gene encodes a protein essential for melanophore development and melanin distribution, influencing variations in human skin pigmentation. This protein plays a vital role in transporting sodium, calcium, and potassium ions across cell membranes, thereby maintaining ion homeostasis and regulating cellular processes [36,37,38].
To investigate the biological functions of the genes identified as model-specific under natural selection by the three PBS models, we conducted Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) pathway enrichment analyses on sets of 36, 53, and 32 genes, respectively. These analyses revealed multiple pathways significantly enriched with signals of natural selection. We analyzed the top seven clusters, and their representative enriched terms based on PBS(Tujia−Li−CEU). The heatmap based on enrichment pathway p-values indicated genes associated with trans-synaptic signaling, regulation of neuron projection development, glycerolipid metabolic process, regulation of RNA splicing, cell population proliferation, bone development, and regulation of membrane potential (Fig. 5b and Table S7a). Additionally, we conducted KEGG and GO pathway enrichment analyses on the gene sets selected by PBS(Tujia−Shui−CEU) and PBS(Tujia−Zhuang−CEU). We found genes associated with the regulation of the JNK cascade, regulation of dendrite development, neuronal system, positive regulation of cell projection organization, and other functions (Fig. S6c-f and Table S7b-c).
To comprehensively explore population-specific selection signatures in the Tujia people relative to different geographically neighboring and genetically different ingroup populations, we interactively analyzed the candidate genes in PBS models designating Li, Shui, and Zhuang as ingroups and Europeans as outgroups. We confirmed shared signals of natural selection from the top 0.1% of the PBS distribution across comparisons. Analysis of these candidate regions highlighted eight common genes (ZMYM4, PSMB2, STX8, AGTPBP1, PTPRD, RANBP3L, OAF, and GRIA1) across the three groups (Fig. 5h and Table S8). We identified significant selection on the STX8 gene, encompassing rs9895062 and rs9902579 (Fig. 5c). The STX8 gene encodes Syntaxin 8, a protein implicated in synaptic transmission and intracellular trafficking. The extended haplotype homozygosity (EHH) analysis of approximately 17_9370623_A/G (rs9895062) indicated extended haplotypes for the derived allele G in its upstream and downstream regions (Fig. 5d). The frequency of the derived allele in the Tujia population was 0.77, compared to 0.15 in the TOPMed dataset and 0.73 in the ToMMo dataset. Similarly, the GRIA1 gene, which encodes the Glutamate Ionotropic Receptor AMPA Type Subunit 1 (GluA1), showed significant selection encompassing rs1493395, rs4145160, and rs514336 (Fig. 5e). This receptor plays a crucial role in fast excitatory synaptic transmission in the central nervous system and is key in synaptic plasticity, learning, and memory. EHH analysis of approximately 5_152911857_A/G (rs1493395) revealed extended haplotypes for the derived allele G in its upstream and downstream regions (Fig. 5f). The derived allele frequency was 0.72 in the Tujia population, compared to 0.23 in the TOPMed dataset and 0.62 in the ToMMo dataset (Fig. S7 and Table S6d). These results suggest that genes related to critical biological processes are vital for establishing stable human populations in distinct ecoregions.
To further investigate the biological consequences of adaptive biological variants in the Tujia population, we examined the potential effects of 104 gene signals identified from three models. A Sankey diagram illustrates the association of 26 SNPs across 22 genes with 24 different traits or diseases based on data from the genome-wide association studies (GWAS) catalog. For example, the EDAR locus (rs3827760 and rs260674) is associated with scalp hair shape and tragus size [39, 40]. The LPP locus (rs1513181) relates to gut microbiota relative abundance [41]. The MAGI2 locus (rs28439811) correlates with COVID-19 severity [42]. The MYO3B locus (rs10176755) pertains to visceral fat accumulation [43]. The MYRF locus (rs2071213 and rs509360) is linked to moyamoya disease and trans fatty acid levels [44, 45]. Lastly, the OAF locus (rs692804) is associated with interleukin-25 levels [46] (Fig. 5g). Future phenomic information and basic experiments are necessary to fully interpret these selection signals.

Signals of positive selection within the Tujia population. a Signatures of natural selection in the Tujia people are illustrated using the population branch statistic (PBS) model based on the Tujia-Li-European trio, represented by Manhattan plots. These plots show confidence levels in selection (vertical axis) against genomic coordinates (horizontal axis), with each dot indicating a single nucleotide polymorphism (SNP). The 99.9th percentiles of the PBS distribution were shown as red lines, PBS values over the 99.9th percentile were marked in red. b Bubble plots of KEGG and GO enrichment for PBS (Tujia-Li-European). Dot size corresponded to the number of genes within each pathway, while the color variation signified different log10 P values and represented the pathway signal strength. c, e Zoom-loci results reveal genes that are highly differentiated within the Tujia population, indicating areas of significant genetic divergence. d, f The extended haplotype homozygosity (EHH) around rs9895062 (17_9370623) and rs1493395 (5_152911857). g A Sankey diagram displays the association of 26 SNPs across 22 genes with 24 different traits or diseases, demonstrating the genetic linkages based on three PBS models. h Venn diagrams depict the signatures of selection unique to or shared among the Tujia people as inferred from various statistical analyses
Discussion
Multidisciplinary archaeological and genetic evidence supports the hypothesis that the origin and diversification of ST languages, along with the development and expansion of two Neolithic cultures, may be attributed to rapid demographic growth and the spread of agriculture. Specifically, the dispersal of STs appears to be linked to the geographical expansion of millet agriculture from northern China, particularly from the YRB, towards the west and south along the periphery of the Tibetan Plateau, commencing around 5,000 years ago [12]. Additionally, previous studies have suggested the widespread occurrence of admixture events in the history of most human populations [6, 7, 9,10,11, 47, 48]. In terms of genetic connections, population genetic analysis using forensic genetic markers, such as STRs and Indels, has revealed a genetic affinity between geographically distinct Tujia populations [17, 19, 21]. Moreover, phylogenetic relationships, reconstructed from genetic variations in Y-STRs, indicate a closer genetic relationship between the Tujia and Miao populations than between the Tujia and other groups such as the Dong and Yao [18].
In this study, the fine-scale genetic structure, migration history, natural selection, and medical relevance of the Tujia people were presented. Genomic analysis was conducted on approximately 700,000 genome-wide SNPs from 18 Tujia individuals from Wendou Town, Hubei Province, and the data were integrated with publicly available datasets from three geographically diverse populations: 51 JKT and 19 YHT from Guizhou Province and 29 QJT from Chongqing Province. Analyses including PCA, ADMIXTURE, Fst genetic matrix, and qpWave revealed relative genetic homogeneity among the four Tujia populations compared to other East Asians. With three genetic clusters evident among East Asians in PCA plots, where the Tujia populations occupied an intermediate position between the northern and southern clusters, showing partial overlap with other Sinitic-speaking groups. Genetic affinity was notably strong between the WDT and three other geographically distinct Tujia groups as evidenced by PCA, Fst genetic matrix, IBD estimation, and effective population size analyses among 12 geographically diverse Tibeto-Burman-speaking populations. Furthermore, TreeMix and f3(Sinitic-/HM-speaking populations, WDT; Mbuti) demonstrated a close genetic relationship between the Tujia populations and other Sinitic- and HM-speaking populations, particularly those from southern China. The WDT exhibited a pronounced genetic affinity with the nearby HM-speaking Miao population, suggesting potential genetic admixture, as indicated by significant positive f4 values f4(HM-speaking populations, East Asians; WDT, Mbuti). Admixture history in the WDT was quantified using qpAdm, suggesting that their ancestry can be modeled as simple two-way admixtures, with modern northern sources contributing between 28% and 71.6% of their ancestry and the remainder deriving from modern Southeast Asians. The admixture-f3 and f4 statistics provided further evidence that the Tujia people derive ancestry from both northern and southern East Asia, underscoring complex historical interactions across regional populations.
Recent human evolution has been marked by significant shifts in dietary habits, health conditions and societal structures, which have exerted complex and widespread selection pressures on human populations. The genetic basis underlying these intricate human characteristics has been greatly elucidated by GWASs performed on contemporary populations, uncovering numerous genetic variants associated with phenotypes of interest [49,50,51]. Considering including different ingroups in PBS model designs has the power to identify population-specific or new candidate selection signatures in the previous studies focused on Li people in Hainan island, Lahu, Dai in Yunnan and other ethno-linguistically diverse Chinese populations [6, 9, 52,53,54,55], we used multiple PBS models to explore population-specific selection signatures in the meta-Tujia people. In an effort to discern biological adaptive signatures among the Tujia people, the PBS was calculated using Li/Shui/Zhuang as the ingroup reference and Europeans as the outgroup. Notably, several genes relevant to specific phenotypes were identified, including the SLC24A5 gene, linked to skin pigmentation, and the EDAR gene, associated with variations in scalp hair thickness and tooth morphology. Research has demonstrated that a particular variant of the SLC24A5 gene is predominantly associated with lighter skin pigmentation across various populations and accounts for a considerable proportion of the pigmentary difference between African and European ancestries [56]. Although melanin is essential for the coloration in skin, hair, and eyes, it is imperative to recognize that SLC24A5 gene is only one of multiple genes affecting skin color. Other genes, such as MC1R, TYR, and OCA2 gene also playing significant roles in the complex genetic landscape of human pigmentation. Further analysis revealed genes under positive selection in the Tujia related to nervous system development, cell projection organization, and endosomal transport. Additionally, regional Tujia-specific genetic signals involving genes such as ZMYM4, PSMB2, STX8, AGTPBP1, PTPRD, RANBP3L, OAF and GRIA1 gene were confirmed, underscoring the unique adaptive genetic architecture of this group. One point warrants closer attention in the integrative analysis of the Tujia people from four geographically distinct populations. Although qpWave results indicated that the Tujia are more homogeneous compared to other minority groups, they do not uniformly share the most derived alleles among themselves. Instead, they also share these alleles with geographically proximate Han Chinese populations. This finding aligns with the extensive genetic admixture signals observed in this study. Consequently, the reported selection signatures may also be present in nearby Han Chinese populations. This hypothesis should be further validated through a large-scale whole-genome sequencing project.
Conclusions
Genetic clustering, utilizing the allele frequency spectrum and shared IBD, revealed relative genetic homogeneity among four geographically diverse Tujia populations, demonstrating a genetic affinity with neighboring Han Chinese and some of the HM-speaking Miao populations. Admixture models clearly indicated that the WDTs possess northern ancestry, along with additional gene flows from SEAs, suggesting an admixture history that incorporates genetic contributions from surrounding regions following their separation from the common Sino-Tibetan ancestor. These findings confirmed distinct regional Tujia signals, reflecting the differentiated adaptation processes across geographically diverse populations.
Materials and methods
Sample collection and ethical statement
We collected saliva samples from 18 unrelated Tujia individuals in Wendou town, Hubei Province. Prior to participation, all subjects provided informed consent in accordance with the Declaration of Helsinki. The study protocol received approval from the Medical Ethics Committees of West China Hospital, Sichuan University (2023 − 306). All participants were confirmed to have parents and grandparents from the same ethnic group, with a history of nonconsanguineous marriages spanning at least three generations.
DNA sequencing, genotyping, and quality control
We extracted and purified DNA utilizing the PureLink Genomic DNA Mini Kit and genotyped genome-wide SNP data via the Illumina array, resulting in the acquisition of approximately 700,000 SNPs across the entire genome in 18 individuals. For quality control filtering, we used PLINK 1.9 [57] with parameter settings of mind: 0.01, geno: 0.01, --hwe 0.001, and --maf 0.01 [57]. Additionally, we assessed family relationships by estimating kinship coefficients using KING2.
Data merging
We combined our data from 18 WDT samples with publicly available data from three geographically diverse populations, including 51 JKT and 19 YHT from Guizhou Province and 29 QJT from Chongqing Province. We categorized these populations as Tujia and merged them with data from previously published present-day and ancient populations [12, 58]. These data were included in the Human Origins (HO) dataset and the 1240 K dataset from the Reich Laboratory, accessible at Reich Lab datasets, along with genomic resources from the Human Genome Diversity Project (HGDP) and Oceania [59,60,61] (Fig. 1a and Table S1). Additionally, data from 5,508 samples previously genotyped in our laboratory using an Illumina chip were integrated into this analysis [6, 62,63,64]. We created three principal merged datasets: the low-density HO_Illumina dataset comprising 18,525 samples with 55,649 SNPs, the middle-density 1240K_Illumina dataset containing 15,962 samples with 146,802 SNPs, and the high-density HGDP_Oceania dataset, which includes 1,378 samples with 460,678 SNPs.
Principal component analysis (PCA)
To explore the clustering patterns among the studied and reference populations, we initially performed PCA using the smartPCA program from the EIGENSOFT v.6.1.4 package [65], applied to an Illumina dataset that includes 123 modern populations and 107 ancient populations. We refined our dataset by pruning with the parameters “-indep-pairwise 200 25 0.4” using PLINK 1.9 to eliminate strongly linked SNPs. To further delineate the fine-scale population structure, we conducted a subsequent PCA on a merged, high-density Illumina dataset that comprises 63 modern East Asian populations and 12 TB-speaking populations at various altitudes. We visualized the results by generating a scatter diagram using R version 4.2.2, enhanced by our in-house scripts.
Model-based unsupervised ADMIXTURE
We integrated our data with a diverse array of both modern and ancient reference populations to investigate the genetic composition of the Tujia individuals. For this analysis, we used the ADMIXTURE software in unsupervised mode, applying the default tenfold cross-validation (--cv = 10) to evaluate the genetic ancestry [66]. We addressed linkage disequilibrium using PLINK 1.9, setting the parameters to r2 > 0.4 and --indep-pairwise 200 25 0.4 to ensure robust data integrity. We then implemented the admixture models across a range of predefined ancestral sources from 2 to 20 (K = 2 ~ 20), conducting 100 bootstraps with various random seeds to assess consistency and reliability. The optimal models were determined by selecting those with the lowest cross-validation error estimates, ensuring the most accurate representation of ancestral genetic contributions.
Fst estimates and TreeMix
We calculated the Fst genetic matrix [67] using an in-house script and PLINK 1.9 to assess the genetic similarities and differences between the Tujia population and other modern East Asian groups [57]. Furthermore, we examined the phylogenetic relationships based on allele frequency data using unsupervised clustering analysis of TreeMix version 1.13 with the default parameters to generate the best-fitted model with migration events ranging from 0 to 19 among 48 modern populations [68]. We also analyzed shared haplotype patterns using MEGA version 7.0 with default settings [69].
Allele-based shared ancestry estimation
We conducted multiple analyses using ADMIXTOOLS to investigate the genetic structure and admixture history of the Tujia people [59]. Initially, we used the qp3pop package, using default parameters and a block jackknife, to compute outgroup-f3 statistics f3(source 1, WDT; Mbuti), assessing shared genetic drift among 63 modern and 49 ancient East Asian populations. Subsequently, we performed admixture-f3 statistics in f3(source 1, source 2; targeted population) to identify admixture signals in Tujia samples from various modern and ancient East Asian source candidates. We identified significant admixture when negative f3 values were accompanied by Z scores less than − 3, indicating that the targeted population was a mixture of two parental populations, source 1 and source 2. Furthermore, we applied f4 statistics in the form of f4(W, X; Y, outgroup) to evaluate potential admixture signals from diverse source populations into the targeted populations. For this, we used the qpDstat package in ADMIXTOOLS with an additional parameter f4Mode (f4: YES) and implemented the block jackknife method to estimate standard errors.
qpWave/qpAdm estimation
We used the qpWave/qpAdm programs from ADMIXTOOLS [59] using default parameters to identify the minimum number of ancestral populations and to calculate the admixture proportions. To enhance the reliability of our estimated admixture models, we incorporated a diverse set of outgroups, including Mbuti, Ust_Ishim, Kostenki14, GoyetQ116_1, Villabruna, Natufian, Atayal, Mixe, Onge, and Cambodian. Additionally, we utilized the “allsnps: YES” parameter to ensure comprehensive SNP inclusion in the analysis.
IBD estimation and effective population size
We utilized refined IBD software (version 16May19. ad5. jar) with default parameter settings to estimate individual pairwise IBDs [70, 71]. Subsequently, we calculated the average total IBD among populations based on the length of individual IBD segments, distinguishing between those ranging from one to five and those over five, reflecting ancient genetic interactions occurring between 500 ~ 1500 years ago and those within the last 500 years, respectively. We also estimated the number and length of shared IBD fragments among 12 TB-speaking populations. Finally, the IBDNe was used to estimate the effective population size among geographically diverse TB-speaking populations over the last 150 generations [72].
Identification of natural selection signals
We used the PBS to identify genomic regions undergoing recent positive selection in the Tujia population, aiming to characterize population-specific variants [67]. The PBS is a robust method that quantifies genetic differentiation between populations and assists in identifying variants under positive selection compared to other populations. Our analysis followed two main steps:
(1) PBS calculation: We calculated the PBS for each genetic variant using allele frequency data from the Tujia population as our target, and two reference populations. The formula used for PBS calculation is PBSA = (TAB+TAC−TBC)/2, where T = − log(1 − FST) [73], A represents the target population, and B and C denote the ingroup and outgroup reference populations, respectively. The FST values measure genetic differentiation between populations. Following the previous studies aimed at identifying signatures in different time-scale branches [6, 9, 52,53,54,55], we conducted multiple PBS models with different ingroug reference populations to identify possible candidates loci or genes.
(2) Selection of candidate variants: After calculating the PBS values for all variants, we identified candidates for positive selection by selecting those within the top 0.1% of PBS values, and the mutation site of the gene was present at least twice. This stringent threshold was chosen to ensure focus on the most extreme outliers, which are more likely to represent true signals of selection. For our study, we consolidated the four geographically diverse Tujia groups into a single population due to their substantial homogeneity. We used Tujia as the target population, with European as the outgroup reference population, and Li_Hainan/Shui_Guizhou and Zhuang_Guangxi as ingroup reference populations.
To further explore region-specific adaptive signals and assess medical associations between adaptive variants and traits, we utilized the GWAS catalog. Additionally, we conducted GO and KEGG enrichment analyses using the online tool Metascape [74] and R version 4.2.2, facilitating the generation of an enrichment dot bubble chart to visually depict our findings.
Data availability
No datasets were generated or analysed during the current study.
References
Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Paabo S. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci U S A. 2013;110(6):2223–7.
Zhong H, Shi H, Qi XB, Xiao CJ, Jin L, Ma RZ, Su B. Global distribution of Y-chromosome haplogroup C reveals the prehistoric migration routes of African exodus and early settlement in East Asia. J Hum Genet. 2010;55(7):428–35.
Li X, Wang M, Su H, Duan S, Sun Y, Chen H, Wang Z, Sun Q, Yang Q, Chen J et al. Evolutionary history and biological adaptation of Han Chinese people on the Mongolian Plateau. hLife 2024.
He G, Wang M, Luo L, Sun Q, Yuan H, Lv H, Feng Y, Liu X, Cheng J, Bu F et al. Population genomics of Central Asian peoples unveil ancient Trans-Eurasian genetic admixture and cultural exchanges. hLife 2024.
Wang M, Chen H, Luo L, Huang Y, Duan S, Yuan H, Tang R, Liu C, He G. Forensic investigative genetic genealogy: expanding pedigree tracing and genetic inquiry in the genomic era. J Genet Genomics 2024.
Sun Y, Wang M, Sun Q, Liu Y, Duan S, Wang Z, et al. Distinguished biological adaptation architecture aggravated population differentiation of Tibeto-Burman-speaking people. J Genet Genomics. 2024;51(5):517–30.
Wang M, Huang Y, Liu K, Wang Z, Zhang M, Yuan H, et al. Multiple human population movements and cultural dispersal events shaped the landscape of Chinese paternal heritage. Mol Biol Evol. 2024;41(7).
Zhang M, Yan S, Pan W, Jin L. Phylogenetic evidence for Sino-Tibetan origin in northern China in the late neolithic. Nature. 2019;569(7754):112–5.
Sun Q, Wang M, Lu T, Duan S, Liu Y, Chen J, Wang Z, Sun Y, Li X, Wang S, et al. Differentiated adaptative genetic architecture and language-related demographical history in South China inferred from 619 genomes from 56 populations. BMC Biol. 2024;22(1):55.
He GL, Wang PX, Chen J, Liu Y, Sun YT, Hu R, Duan SH, Sun QX, Tang RK, Yang JB, et al. Differentiated genomic footprints suggest isolation and long-distance migration of Hmong-Mien populations. BMC Biol. 2024;22(1):18.
He G, Wang M, Miao L, Chen J, Zhao J, Sun Q, Duan S, Wang Z, Xu X, Sun Y, et al. Multiple founding paternal lineages inferred from the newly-developed 639-plex Y-SNP panel suggested the complex admixture and migration history of Chinese people. Hum Genomics. 2023;17(1):29.
Wang CC, Yeh HY, Popov AN, Zhang HQ, Matsumura H, Sirak K, Cheronet O, Kovalev A, Rohland N, Kim AM, et al. Genomic insights into the formation of human populations in East Asia. Nature. 2021;591(7850):413–9.
He G, Wang M, Zou X, Chen P, Wang Z, Liu Y, Yao H, Wei LH, Tang R, Wang CC, et al. Peopling history of the Tibetan Plateau and multiple waves of admixture of tibetans inferred from both ancient and modern genome-wide data. Front Genet. 2021;12(1634):725243.
He G, Li YX, Wang MG, Zou X, Yeh HY, Yang XM, Wang Z, Tang RK, Zhu SM, Guo JX, et al. Fine-scale genetic structure of Tujia and central Han Chinese revealing massive genetic admixture under language borrowing. J Syst Evol. 2021;59(1):1–20.
Wang CC, Wang LX, Shrestha R, Zhang M, Huang XY, Hu K, Jin L, Li H. Genetic structure of Qiangic populations residing in the western Sichuan corridor. PLoS ONE. 2014;9(8):e103772.
Chen FH, Dong GH, Zhang DJ, Liu XY, Jia X, An CB, Ma MM, Xie YW, Barton L, Ren XY, et al. Agriculture facilitated permanent human occupation of the Tibetan Plateau after 3600 B.P. Science. 2015;347(6219):248–50.
Lan J, Zhang X, Cui W, Mei S, Xu J, Zhu B. Genetic polymorphisms and population genetic analyses of 57 autosomal InDel loci in Hubei Tujia group. Front Genet. 2023;14:1066655.
Feng R, Zhao Y, Chen S, Li Q, Fu Y, Zhao L, Zhou Y, Zhang L, Mei X, Shi M, et al. Genetic analysis of 50 Y-STR loci in Dong, Miao, Tujia, and Yao populations from Hunan. Int J Legal Med. 2020;134(3):981–3.
Zhang L, Yang F, Bai X, Yao Y, Li J. Genetic polymorphism analysis of 23 STR loci in the Tujia population from Chongqing, Southwest China. Int J Legal Med. 2021;135(3):761–3.
Luo L, Gao H, Yao L, Long F, Zhang H, Zhang L, Liu Y, Yu J, Yu L, Chen P. Genetic diversity, forensic feature, and phylogenetic analysis of Guizhou Tujia population via 19 X-STRs. Mol Genet Genomic Med. 2020;8(11):e1473.
Shen C, Zhu B, Yao T, Li Z, Zhang Y, Yan J, Wang B, Bie X, Tai F. A 30-InDel assay for Genetic Variation and Population Structure Analysis of Chinese Tujia Group. Sci Rep. 2016;6:36842.
He G, Li Y, Wang M, Zou X, Yeh H, Yang X, et al. Fine-scale genetic structure of Tujia and central Han Chinese revealing massive genetic admixture under language borrowing. J Syst Evol. 2021;59(1):1–20.
He GL, Wang MG, Zou X, Yeh HY, Liu CH, Liu C, Chen G, Wang CC. Extensive ethnolinguistic diversity at the crossroads of North China and South Siberia reflects multiple sources of genetic diversity. J Syst Evol. 2022;61(1):230–50.
Lipson M, Loh PR, Patterson N, Moorjani P, Ko YC, Stoneking M, Berger B, Reich D. Reconstructing austronesian population history in Island Southeast Asia. Nat Commun. 2014;5:4689.
Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo JM, Lema G, Fu W, Nyambo TB, Rebbeck TR, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150(3):457–69.
Pagani L, Lawson DJ, Jagoda E, Mörseburg A, Eriksson A, Mitt M, Clemente F, Hudjashov G, DeGiorgio M, Saag L, et al. Genomic analyses inform on migration events during the peopling of Eurasia. Nature. 2016;538(7624):238–42.
Malaspinas AS, Westaway MC, Muller C, Sousa VC, Lao O, Alves I, Bergström A, Athanasiadis G, Cheng JY, Crawford JE, et al. A genomic history of Aboriginal Australia. Nature. 2016;538(7624):207–14.
Mondal M, Casals F, Xu T, Dall’Olio GM, Pybus M, Netea MG, Comas D, Laayouni H, Li Q, Majumder PP, et al. Genomic analysis of Andamanese provides insights into ancient human migration into Asia and adaptation. Nat Genet. 2016;48(9):1066–70.
Mallick S, Micco A, Mah M, Ringbauer H, Lazaridis I, Olalde I, Patterson N, Reich D. The Allen Ancient DNA resource (AADR) a curated compendium of ancient human genomes. Sci Data. 2024;11(1):182.
Yang X, Sarengaowa, He G, Guo J, Zhu K, Ma H, Zhao J, Yang M, Chen J, Zhang X, et al. Genomic insights into the genetic structure and natural selection of mongolians. Front Genet. 2021;12:735786.
Skoglund P, Thompson JC, Prendergast ME, Mittnik A, Sirak K, Hajdinjak M, et al. Reconstructing prehistoric African population structure. Cell. 2017;171(1):59–71, e21.
André M, Brucato N, Hudjasov G, Pankratov V, Yermakovich D, Montinaro F, Kreevan R, Kariwiga J, Muke J, Boland A, et al. Positive selection in the genomes of two Papua New Guinean populations at distinct altitude levels. Nat Commun. 2024;15(1):3352.
Fujimoto A, Ohashi J, Nishida N, Miyagawa T, Morishita Y, Tsunoda T, Kimura R, Tokunaga K. A replication study confirmed the EDAR gene to be a major contributor to population differentiation regarding head hair thickness in Asia. Hum Genet. 2008;124(2):179–85.
Fujimoto A, Kimura R, Ohashi J, Omi K, Yuliwulandari R, Batubara L, Mustofa MS, Samakkarn U, Settheetham-Ishida W, Ishida T, et al. A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum Mol Genet. 2008;17(6):835–43.
Williams R, Jobling S, Sims AH, Mou C, Wilkinson L, Collu GM, Streuli CH, Gilmore AP, Headon DJ, Brennan K. Elevated EDAR signalling promotes mammary gland tumourigenesis with squamous metaplasia. Oncogene. 2022;41(7):1040–9.
Reis LB, Bakos RM, Vianna FSL, Macedo GS, Jacovas VC, Ribeiro-Dos-Santos AM, Santos S, Bakos L, Ashton-Prolla P. Skin pigmentation polymorphisms associated with increased risk of melanoma in a case-control sample from southern Brazil. BMC Cancer. 2020;20(1):1069.
Giardina E, Pietrangeli I, Martínez-Labarga C, Martone C, de Angelis F, Spinella A, De Stefano G, Rickards O, Novelli G. Haplotypes in SLC24A5 gene as Ancestry informative markers in different populations. Curr Genom. 2008;9(2):110–4.
Basu Mallick C, Iliescu FM, Mols M, Hill S, Tamang R, Chaubey G, Goto R, Ho SY, Gallego Romero I, Crivellaro F, et al. The light skin allele of SLC24A5 in South asians and europeans shares identity by descent. PLoS Genet. 2013;9(11):e1003912.
Adhikari K, Reales G, Smith AJ, Konka E, Palmen J, Quinto-Sanchez M, Acuña-Alonzo V, Jaramillo C, Arias W, Fuentes M, et al. A genome-wide association study identifies multiple loci for variation in human ear morphology. Nat Commun. 2015;6:7500.
Adhikari K, Fontanil T, Cal S, Mendoza-Revilla J, Fuentes-Guajardo M, Chacon-Duque JC, Al-Saadi F, Johansson JA, Quinto-Sanchez M, Acuna-Alonzo V, et al. A genome-wide association scan in admixed Latin americans identifies loci influencing facial and scalp hair features. Nat Commun. 2016;7:10815.
Ishida S, Kato K, Tanaka M, Odamaki T, Kubo R, Mitsuyama E, Xiao JZ, Yamaguchi R, Uematsu S, Imoto S, et al. Genome-wide association studies and heritability analysis reveal the involvement of host genetics in the Japanese gut microbiota. Commun Biol. 2020;3(1):686.
Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, Walker S, Parkinson N, Fourman MH, Russell CD, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–8.
Fox CS, Liu Y, White CC, Feitosa M, Smith AV, Heard-Costa N, Lohman K, Johnson AD, Foster MC, Greenawalt DM, et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012;8(5):e1002695.
Mozaffarian D, Kabagambe EK, Johnson CO, Lemaitre RN, Manichaikul A, Sun Q, Foy M, Wang L, Wiener H, Irvin MR, et al. Genetic loci associated with circulating phospholipid trans fatty acids: a meta-analysis of genome-wide association studies from the CHARGE Consortium. Am J Clin Nutr. 2015;101(2):398–406.
Duan L, Wei L, Tian Y, Zhang Z, Hu P, Wei Q, Liu S, Zhang J, Wang Y, Li D, et al. Novel susceptibility loci for Moyamoya Disease revealed by a genome-wide Association study. Stroke. 2018;49(1):11–8.
Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, DeLisle RK, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357.
Chen J, He G, Ren Z, Wang Q, Liu Y, Zhang H, et al. Genomic Insights Into the Admixture History of Mongolic- and Tungusic-Speaking Populations From Southwestern East Asia. Front Genet. 2021;12:685285.
Chen J, He G, Ren Z, Wang Q, Liu Y, Zhang H, Yang M, Zhang H, Ji J, Zhao J, et al. Fine-Scale Population Admixture Landscape of Tai-Kadai-speaking Maonan in Southwest China inferred from genome-wide SNP data. Front Genet. 2022;13:815285.
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 years of GWAS Discovery: Biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–12.
Yang Z, Chen H, Lu Y, Gao Y, Sun H, Wang J, Jin L, Chu J, Xu S. Genetic evidence of tri-genealogy hypothesis on the origin of ethnic minorities in Yunnan. BMC Biol. 2022;20(1):166.
Liu C, Yuan B, Wang D, Xu J, Wang M. Genome-wide allele and haplotype-sharing patterns suggested one unique Hmong-Mein-related lineage and biological adaptation history in Southwest China. Hum Genomics. 2023;17(1):3.
Chen H, Lin R, Lu Y, Zhang R, Gao Y, He YG, Xu SH. Tracing Bai-Yue Ancestry in Aboriginal Li people on Hainan Island. Mol Biol Evol. 2022;39(10):msac210.
Su H, Wang M, Li X, Duan S, Sun Q, Sun Y, et al. Population genetic admixture and evolutionary history in the Shandong Peninsula inferred from integrative modern and ancient genomic resources. BMC Genomics. 2024;25(1):611.
Yang Z, Zhong H, Chen J, Zhang X, Zhang H, Luo X, Xu S, Chen H, Lu D, Han Y, et al. A genetic mechanism for convergent skin lightening during recent human evolution. Mol Biol Evol. 2016;33(5):1177–87.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Ning C, Li T, Wang K, Zhang F, Li T, Wu X, Gao S, Zhang Q, Zhang H, Hudson MJ, et al. Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat Commun. 2020;11(1):2700.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. Ancient admixture in human history. Genetics. 2012;192(3):1065–93.
Bergström A, McCarthy SA, Hui R, Almarri MA, Ayub Q, Danecek P, Chen Y, Felkel S, Hallast P, Kamm J et al. Insights into human genetic variation and population history from 929 diverse genomes. Science. 2020;367(6484).
Choin J, Mendoza-Revilla J, Arauna LR, Cuadros-Espinoza S, Cassar O, Larena M, Ko AM, Harmant C, Laurent R, Verdu P, et al. Genomic insights into population history and biological adaptation in Oceania. Nature. 2021;592(7855):583–9.
He GL, Li YX, Zou X, Yeh HY, Tang RK, Wang PX, Bai JY, Yang XM, Wang Z, Guo JX, et al. Northern gene flow into southeastern East asians inferred from genome-wide array genotyping. J Syst Evol. 2022;61(1):179–97.
He GL, Wang MG, Li YX, Zou X, Yeh HY, Tang RK, Yang XM, Wang Z, Guo JX, Luo T, et al. Fine-scale north‐to‐south genetic admixture profile in Shaanxi Han Chinese revealed by genome‐wide demographic history reconstruction. J Syst Evol. 2021;60(4):955–72.
Wang MG, He GL, Zou X, Chen PY, Wang Z, Tang RK, Yang XM, Chen J, Yang MQ, Li YX, et al. Reconstructing the genetic admixture history of Tai-Kadai and sinitic people: insights from genome‐wide SNP data from South China. J Syst Evol. 2022;61(1):157–78.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64.
Weir BS, Cockerham CC. Estimating F-Statistics for the analysis of Population structure. Evolution. 1984;38(6):1358–70.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Browning BL, Browning SR. A fast, powerful method for detecting identity by descent. Am J Hum Genet. 2011;88(2):173–82.
Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics. 2013;194(2):459–71.
Browning SR, Browning BL. Accurate non-parametric estimation of recent effective Population size from segments of identity by Descent. Am J Hum Genet. 2015;97(3):404–18.
Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329(5987):75–8.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Acknowledgements
We thank Prof. Mark Stoneking, Prof. Dang Liu at the Max Planck Institute for Evolutionary Anthropology, and Prof. Wibhu Kutanan at Khon Kaen University for sharing genome-wide SNP data from Vietnam, Thailand, and Laos. We thank Prof. Etienne Patin and Prof. Lluis Quintana-Murci from the Human Evolutionary Genetics Unit of the Institute Pasteur for sharing the high-coverage genomes of 317 individuals from the Pacific region.
Funding
This work was supported by the National Natural Science Foundation of China (82030058), the National Natural Science Foundation of China (82402203 and 82202078), the Major Project of the National Social Science Foundation of China (23&ZD203), the Open Project of the Key Laboratory of Forensic Genetics of the Ministry of Public Security (2022FGKFKT05), the Center for Archaeological Science of Sichuan University (23SASA01), the 1‧3‧5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC20002), and the Sichuan Science and Technology Program (2024NSFSC1518) and the Shanxi Province Graduate Education Innovation Program Project (2023KY369).
Author information
Authors and Affiliations
Contributions
J.Y. and G.H. conceived and supervised the project. G.H., Y.L., and S.D. collected the samples. G.H., M.W., and Y.L. extracted the genomic DNA and coordinated the genome sequencing. J.C., S.D., M.Z., Q.S., X.L., Y.S., H.S., Q.Y., Z.W., Y.H., J.Z., Y.F., and X.Z. performed the population genetic analysis. J.C., M.W., and G.H. drafted the manuscript. J.Y., G.H. and M.W. revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Informed consent
Informed consent was obtained from all subjects involved in the study.
Institutional review board statement
The Medical Ethics Committees of West China Hospital of Sichuan University approved this study. This study adhered to the principles of the Helsinki Declaration.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40246_2024_672_MOESM1_ESM.tif
Supplementary Material 1: Fig. S1. Detailed legends for Fig. 1b-d
40246_2024_672_MOESM2_ESM.tif
Supplementary Material 2: Fig. S2. Results of model-based ADMIXTURE clustering analysis among 230 East Asian populations at K = 8.
40246_2024_672_MOESM3_ESM.tif
Supplementary Material 3: Fig. S3. Genetic affinity between Tujia populations and modern East Asians. a Principal component analysis between Tujia and other modern references of East Asians based on the 1240k dataset. The genetic background was constructed based on the genetic variations from modern populations and their top two components. Modern populations were color-coded based on their language family categories. b Principal component analysis between the WDT and Tibeto-Burman-speaking populations at 11 different altitudes. The red circle highlights four Tujia people. c Results of model-based ADMIXTURE clustering analysis among 85 populations in East Asia at K = 5.
40246_2024_672_MOESM5_ESM.tif
Supplementary Material 5: Fig. S5. The best-fitted model with migration events ranging from 0 to 19 among 48 modern populations.
40246_2024_672_MOESM6_ESM.tif
Supplementary Material 6: Fig. S6. Manhattan plots and Enrichment analysis of PBS (Tujia−Shui−European) and PBS (Tujia−Zhuang−European) biological adaptive signals. a Signatures of natural selection identified in the Tujia people based on the PBS values in the Tujia-Shui_Guizhou-European trio model. b Signatures of natural selection identified in the Tujia people based on the PBS values in the Tujia-Zhuang_Guangxi-European trio model. c Heatmap of the gene list for the top 11 enriched terms of PBS (Tujia−Shui−European), colored by P value. d Heatmap of the gene list for the top 8 enriched terms of PBS (Tujia−Zhuang−European), colored by P value. e Bubble plots of KEGG and GO enrichment for PBS (Tujia−Shui−European). f Bubble plots of KEGG and GO enrichment for PBS (Tujia−Zhuang−European).
40246_2024_672_MOESM7_ESM.tif
Supplementary Material 7: Fig. S7. a Zoom-loci results reveal genes that are highly differentiated within the Tujia population, indicating areas of significant genetic divergence. b The EHH around rs9902579 (17_9352325), rs9895062 (17_9370623), rs2133766 (5_36278890), rs1551240 (11_120087541), rs56667128 (11_120097175), rs1493395 (5_152911857), rs514336(5_152902390), and rs4145160 (5_152874549).
40246_2024_672_MOESM8_ESM.xls
Supplementary Material 8: Table S1. Detailed information of the included populations in the merged Human Origins dataset used for model-based analysis.
40246_2024_672_MOESM9_ESM.xls
Supplementary Material 9: Table S2. a Differentiated shared alleles between the other three published Tujia groups and WDT compared to modern East Asians, estimated via symmetrical f4 statistics in the form of f4 (JKT/QJT/YHT, WDT; modern East Asians, Mbuti) based on the merged Human Origins dataset. b Differentiated shared alleles between the other three published Tujia groups and WDT compared to ancient East Asians, estimated via symmetrical f4 statistics in the form of f4 (JKT/QJT/YHT, WDT; ancient East Asians, Mbuti) based on the merged Human Origins dataset. c Differentiated shared alleles between JKT and the other three Tujia groups compared to modern East Asians, estimated via symmetrical f4 statistics in the form of f4 (JKT, WDT/QJT/YHT; East Asians, Mbuti).
40246_2024_672_MOESM10_ESM.xls
Supplementary Material 10: Table S3. a Affinity-f4 statistics in the form of f4 (high-altitude ST-speaking populations, East Asians; WDT, Mbuti) based on the merged Human Origins dataset. b Affinity-f4 statistics in the form of f4 (Hmong-Mien-speaking populations, East Asians; WDT, Mbuti) based on the merged human origins dataset. c Affinity-f4 statistics in the form of f4 (Blang/Wa, East Asians; WDT, Mbuti) based on the merged Human Origins dataset. d Affinity-f4 statistics in the form of f4 (Gaoshan, East Asians; WDT, Mbuti) based on the merged Human Origins dataset. e Affinity-f4 statistics in the form of f4 (Tai-Kadai-speaking populations, East Asians; WDT, Mbuti) based on the merged Human Origins dataset. f Affinity-f4 statistics in the form of f4 (Hmong-Mien-speaking populations, Tai-Kadai-speaking populations; WDT, Mbuti) based on the merged human origins dataset.
40246_2024_672_MOESM11_ESM.xls
Supplementary Material 11: Table S4. a Affinity-f4 statistics in the form of f4 (Baojianshan/Longlin, ancient East Asians; WDT, Mbuti). b Nonsymmetrical f4 statistics in the form of f4 (ancient East Asians, WDT; YRB ancients, Mbuti). c Nonsymmetrical f4 statistics in the form of f4 (ancient ANEAs, WDT; ancient East Asians, Mbuti).
40246_2024_672_MOESM13_ESM.xls
Supplementary Material 13: Table S6. a The top 0.01% of signals were selected by PBS (Tujia−Li−CEU). b The top 0.01% of signals selected by PBS (Tujia−Shui−CEU). c The top 0.01% of signals were selected by PBS (Tujia−Zhuang−CEU). d Frequency distribution of the eight loci in Tujia and public database.
40246_2024_672_MOESM14_ESM.xls
Supplementary Material 14: Table S7. a Top 7 clusters with their representative enriched terms (one per cluster) based on PBS (Tujia−Li−CEU). b Top 11 clusters with their representative enriched terms (one per cluster) based on PBS(Tujia−Shui−CEU). c Top 8 clusters with their representative enriched terms (one per cluster) based on PBS(Tujia−Zhuang−CEU).
40246_2024_672_MOESM15_ESM.xls
Supplementary Material 15: Table S8. Distinct and shared genes related to biological adaptation in Tujia among the PBS (Tujia−Li−CEU), PBS (Tujia−Shui−CEU) and PBS (Tujia−Zhuang−CEU) groups.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, J., Wang, M., Duan, S. et al. Genetic history and biological adaptive landscape of the Tujia people inferred from shared haplotypes and alleles. Hum Genomics 18, 104 (2024). https://doi.org/10.1186/s40246-024-00672-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-024-00672-y