
Abstract
Since the discovery of group 2 innate lymphoid cells (ILC2s) in 2010, subsequent studies have revealed their developmental pathways, mechanisms of activation and regulation, and immunological roles in tissue homeostasis and tissue-specific diseases in various organs. Although ILC2s are known to express tissue-specific features depending on where they reside, how the surrounding environment affects the functions of ILC2s remains to be fully elucidated. Recent histologic analyses revealed that ILC2s resides in specific perivascular regions in peripheral tissues with their function being controlled by the surrounding cells via cytokines, lipid mediators, neurotransmitters, and cell–cell interactions through surface molecules. This review summarizes the interactions between ILC2s and surrounding cells, including epithelial cells, neurons, immune cells, and mesenchymal cells, with the objective of promoting the development of novel diagnostic and therapeutic methods for ILC2-related diseases.
Similar content being viewed by others


Background
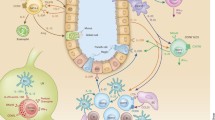
Group 2 innate lymphoid cells (ILC2s) are a subset of innate lymphoid cells that express transcription factor GATA-binding protein 3 (GATA3) and lack specific lineage markers [1]. Unlike adaptive lymphocytes, ILC2s do not possess antigen-recognition receptors such as the T cell antigen receptor or B-cell antigen receptor. Instead of antigen-specific responses, ILC2s respond to environmental factors including cytokines, neuropeptides, lipid mediators, hormones, and nutrients. In response to these inputs, ILC2s produce various cytokines, such as IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, GM-CSF, and amphiregulin. By producing these cytokines, ILC2s are associated with the pathogenesis of various diseases, including parasitic infections, allergic diseases, autoimmune diseases, metabolic diseases, and malignant diseases (Fig. 1) [2,3,4,5,6,7,8,9,10]. ILC2s were first discovered in murine adipose tissue [2] but are now known to be present in various tissues in both humans and mice, for example, lung, intestine, brain, heart, liver, skin, and peripheral blood [11,12,13,14,15,16,17]. ILC2s are generally tissue-resident cells that possess tissue-specific functions. Progress in the past several years has clarified the factors that regulate the activation, suppression, expansion, development, and migration of ILC2s. However, how the surrounding environment affects tissue-resident ILC2s is not fully understood. This review summarizes recent advances in the understanding of mechanisms regulating ILC2s by surrounding cells, which would contribute to the development of basic studies on ILC2s and the investigation of new clinical targets for ILC2-associated disorders.

ILC2s and its related diseases. Studies with human and mouse revealed that ILC2s reside in various tissues exhibiting tissue-specific features and play a pivotal role in maintaining the homeostasis of tissues they reside
Development and migration of ILC2s
ILC precursors and ILC2s first arise during fetal development, both in humans and mice. They are seeded in peripheral tissues and differentiate in situ to acquire tissue-specific profiles [18,19,20]. Shortly after birth, fetal ILC2s are followed by neonatal origin ILC2s, which comprise the majority of adult peripheral ILC2 pools accompanied by the acquisition of effector repertoires and tissue-specific signatures [21, 22].
In adult parabiotic mice, ILC2s were shown to be tissue-resident cells. They are maintained and expanded locally both during homeostatic conditions and acute helminth infection. However, during the chronic phase of infection, a fraction of ILC2s from hematogenous sources are recruited to replenish the pool of resident ILC2s, leading to the clearance of the infection and tissue healing [23]. In addition to helminth infection, IL-25 induces inflammatory ILC2s in the small intestines to proliferate and traffic to the lymphatics and blood circulation. They reach the peripheral tissues, including the lungs, in a sphingosine-1-phosphate-dependent manner [24]. Ricardo-Gonzalez et al. showed that the activation of local ILC2s by tissue-specific alarmins induces ILC2 proliferation, lymph node migration, and blood dissemination, which causes local innate responses to transition into systemic type 2 responses [25]. As mentioned above, ILC2s are basically tissue-resident cells; however, in certain conditions, they migrate from tissue to tissue and they turn local inflammation into systemic inflammation.
Recent studies have shown that tissue niches play a crucial role in the acquisition of tissue-specific properties by tissue-resident or migratory ILC2s. Using a single-cell atlas of lung ILC2s, Zeith et al. showed that tissue-resident Il18r1+ ILC2 precursors contribute to the maintenance and renewal of tissue ILC2s via in situ differentiation, suggesting that local niches, rather than origin or developmental period, might dominantly imprint ILC phenotypes in adult tissues [26]. For instance, neuropilin-1 (Nrp1) is a tissue-specific marker of lung ILC2s that is induced postnatally and sustained by lung-derived transforming growth factor beta-1 (TGFβ1). TGFβ1–Nrp1 signaling enhances ILC2 function and exacerbates bleomycin-induced lung fibrosis through upregulation of IL-33 receptor ST2 expression [27]. As described above, tissue specificity of ILC2s is acquired through signals from the tissue niches where they reside.
Localization of ILC2s
Although ILC2s are present in various peripheral tissues, ILC2s are enriched in mucosal organs, such as the skin, intestines, and lungs. In these tissues, ILC2s localize right below epithelial cells and are activated by IL-33, which is released from damaged epithelial cells due to parasites or allergens and induces eosinophilic inflammation. In addition, IL-25 produced by tuft cells activates ILC2s and induces enhanced mucus production [2, 28, 29]. Three-dimensional imaging technologies have shown that ILC2s reside in perivascular adventitial cuff spaces, which represent the outermost layer of blood vessels, in multiple sights including the lungs, liver, brain meninges, and adipose tissue. ILC2s are localized with dendritic cells, regulatory T cells, and adventitial stromal cells (ASCs), a mesenchymal fibroblast-like subset expressing IL-33 and thymic stromal lymphopoietin (TSLP) [30]. In addition, pulmonary neuroendocrine cells (PNECs) reside close to ILC2s near airway branch points in the lungs and amplify allergic asthma responses by producing calcitonin gene–related peptide (CGRP) [31]. Single-cell RNA sequencing studies have shown that intestinal ILC2s are regulated by neuropeptide alpha-calcitonin gene–related peptide (α-CGRP) [32]. As described above, tissue-resident ILC2s localize with epithelial cells, immune cells, neurons, and mesenchymal cells in peripheral tissues. We will explain the interaction between ILC2s and these cells below.
Epithelial cells and ILC2s
Epithelial cells in the skin and mucosal tissues, including the lungs and intestines, constitute a barrier between the external environment and the underlying mesenchyme. They also respond to external stimuli and interact with immune cells to maintain homeostasis (Fig. 2).

Regulation of ILC2s by epithelial cells. ILC2s reside just below the epithelial cells in mucosal tissues, such as lung, intestine and skin. IL-33, IL-25, and TSLP are released from epithelial cell in response to epithelial damage due to invasion of allergen, helminth and pathogen. These alarmins activate ILC2s and lead to type 2 inflammation
The epithelial cell–derived danger signal mediators IL-33, TSLP, and IL-25 are consistently associated with type 2 immune responses in allergic diseases [33]. Genome-wide association studies (GWAS) have shown an association between allergic diseases and genetic polymorphisms in genes such as TSLP, IL33, and IL1RL1, which encode the IL-33 receptor ST2 [34]. IL-33 is normally found in the nucleus of epithelial cells. When activated or damaged by exposure to parasites or allergen-derived proteases, full-length IL-33 is released from epithelial cells and rapidly processed into a mature active form, which leads to strong ILC2 activation [35]. Although studies have reported that structural cells, including endothelial cells, fibroblasts, smooth muscle cells, and epithelial cells, express IL-33 in various organs, bronchial epithelium is reported to be an important reservoir of IL-33 in the lungs and expression levels are elevated in patients with bronchial asthma [36]. TSLP expression is also higher in the airways of patients with asthma than in those of healthy controls. TSLP levels correlate with Th2 cytokine and chemokine expression and disease severity [37, 38]. TSLP alone mildly activates ILC2s; however, when administered with IL-33, TSLP induces ILC2 corticosteroid resistance by controlling phosphorylation of STAT5 [39]. Because corticosteroid resistance contributes to uncontrolled severe asthma and TSLP upregulates antigen-specific Th2 cell cytokine production through its activity on innate immune cells as well as dendritic cells, T cells, and B cells, a human monoclonal antibody specific for TSLP has been developed [40]. IL-25 is also released from epithelial cells. Allergen provocation induces increased expression of IL-25 and its receptor in the bronchial mucosa and dermis of patients with asthma or atopic dermatitis [41]. In addition, tuft cells, a cell type in the epithelium of the small intestines, express IL-25 to sustain ILC2 homeostasis in a steady state. During helminth infection, IL-25 derived from tuft cells induces IL-13 secretion from ILC2s, which leads to epithelial remodeling by increasing the number of tuft and goblet cells [29]. Moreover, E-cadherin, an adhesion protein expressed by epithelial cells that is responsible for maintaining the integrity of the epithelium, acts as a ligand for KLRG1 and suppresses ILC2 function to avoid excessive ILC2 activation [17]. In inflamed skin lesions of patients with atopic dermatitis, cleavage of E-cadherin might lead to the discontinuation of the inhibitory signal from ILC2s.
Neurons and ILC2s
The immune and nervous systems can communicate using common molecular signaling cues in various organ systems. Recent studies have revealed that ILC2s express receptors for several neurotransmitters, such as vasoactive intestinal polypeptide (VIP), neuromedin U (NMU), CGRP, and norepinephrine (NE). ILC2s are directly activated and regulated by these neurotransmitters produced by various peripheral neurons (Fig. 3).

Regulation of ILC2s by nervous system. Sensory, sympathetic and parasympathetic neurons regulate the function of ILC2 by various neurotransmitters. Inhaled allergen or food intake stimulates sensory neurons to produce VIP in the lung and intestine respectively. VIP induces IL-5 production by ILC2s, which in turn enhances VIP production. During helminth infection, NE from sympathetic neurons inhibit cell proliferations and cytokine production in both intestine and lung ILC2s. NMU from parasympathetic neuron enhances the proliferation and cytokine secretion by ILC2s, while CGRP suppress the proliferation and IL-13 production from ILC2s
The peripheral nervous system is composed of the somatic nervous system, which includes motor and sensory neurons, and the autonomic nervous system, which includes sympathetic and parasympathetic neurons. Sensory, sympathetic, and parasympathetic neurons are reported to regulate ILC2s independently from cytokine stimuli.
Sensory neurons transmit sensory information about the body and internal organs to the central nervous system. The lungs have a dense network of nociceptors expressing sensory neurons, which produce VIP and exacerbate asthma symptoms by activating T cells and ILC2s via the VIP–VIP receptor type 2 axis. IL-5 produced by activated ILC2s enhances VIP production, creating a positive feedback loop between sensory neurons and ILC2s [42]. Sensory neurons in the small intestines also produce VIP when stimulated by nutrient intake and central circadian rhythms, leading to the induction of IL-5 production by ILC2s and contributing to basal eosinophilopoiesis and eosinophil accumulation in tissues [43].
Sympathetic neurons control the fight-or-flight response in stressed, dangerous, or physically active situations. They increase heart rate and promote vasoconstriction to activate physical activity while suppressing intestinal motility and intestinal tract secretion. The sympathetic nervous system includes adrenergic neurons that produce catecholamines, i.e., epinephrine and NE. Catecholamines exert their effect via two classes of adrenergic receptors α (α1, α2) and β (β1, β2, and β3). ILC2s express the β2-adrenergic receptor (β2AR) and colocalize with adrenergic neurons in the intestines. Moriyama et al. reported that the β2AR pathway is a cell-intrinsic negative regulator of ILC2 responses via inhibition of cell proliferation and effector function [44].
Parasympathetic neurons are responsible for stimulation of rest-and-digest or feed-and-breed activities when the body is at rest. They play an antagonistic role to sympathetic neurons. They reduce heart rate, relax blood vessels, and activate digestive activity. The parasympathetic nervous system mainly uses acetylcholine as its neurotransmitter, which is produced by cholinergic neurons. NMU is produced by cholinergic neurons and signals through its receptors neuromedin U receptor 1 (NMUR1) and NMUR2 [45]. Single-cell RNA sequencing and genome-wide transcriptional profiling have revealed that ILC2s selectively express Nmur1. ILC2s in the murine gastrointestinal tract colocalize with cholinergic neurons that express NMU. NMU–NMU1 signaling induces cell activation, proliferation, and secretion of the type 2 cytokines IL-5, IL-9, and IL-13 from ILC2s. Worm products and alarmins directly stimulates mucosal neurons to induce NMU and lead to accelerated expulsion of Nippostrongylus brasiliensis. Lung ILC2s express NUMR1 in the steady state and after IL-25 stimulation; in vivo co-administration of NMU with IL-25 strongly amplifies allergic inflammation [46,47,48]. Of note, NMU induces both smooth muscle contraction [49], which exacerbates asthma, and ILC2-driven inflammation. On the other hand, CGRP released from parasympathetic neurons suppresses the proliferation of ILC2s and IL-13 production by ILC2s and attenuates type 2 inflammation in the lungs and intestines [32, 50, 51].
In summary, the nervous system appears to have two mechanisms to rapidly activate or repress these innate immune cells in order to maintain tissue homeostasis and protect the host against diverse inflammatory stimuli. These findings highlight the importance of neuro–immune crosstalk at mucosal surfaces. The investigation of neuroimmune interactions with ILC2s might lead to greater understanding of the mechanisms of asthma exacerbation by non-antigenic factors such as air pollutants, cold exposure, and psychological stress.
Immune cells and ILC2s
ILC2s interact with various immune cells via cell–cell contact or communication via soluble factors such as cytokines, lipid mediators, and hormones (Fig. 4). ILC2s colocalize with regulatory T (Treg) cells, CD4+ T helper type 2 (Th2) cells, and dendritic cells (DCs) in the perivascular adventitial cuff of peripheral tissues.

Regulation of ILC2s by immune cells. ILC2s interact with various immune cells by cytokines, lipid mediators, and cell–cell contact. ILC2s and Th2 cells compound positive feedback loop driving innate and adaptive immune responses in allergic inflammation and anti-helminth immunity. In visceral adipose tissue, IL-5 and IL-13 produced from ILC2s promote eosinophils and M2 macrophages implicated in metabolic homeostasis
ILC2s drive the initiation of type 2 innate immune responses, leading to the activation of the adaptive immune system driven by Th2 cells. CD4+T cells provide IL-2, which promotes ILC2 proliferation and IL-13 production. ILC2s, in turn, influence the differentiation of naïve CD4+ T cells into a Th2 phenotype in a contact-dependent manner [52]. Moreover, activated ILC2s upregulate the expression of OX40 ligand and programmed death ligand 1, leading to the activation of Th2 cells by upregulating GATA3 expression and cytokine production [53, 54]. Activated Th2 cells produce IL-4 and IL-9, which contribute to ILC2 activation. This positive feedback loop drives both innate and adaptive immune responses. In addition to Th2 cells, basophil- and eosinophil-derived IL-4 enhances ILC2-derived cytokine and chemokine expression, leading to type 2 inflammation [55, 56]. IL-5 produced by ILC2s enhances the proliferation, survival, and recruitment of eosinophils [43]. Eosinophils, in turn, produce IL-4 to stimulate ILC2s, thereby mediating the crosstalk between eosinophils and ILC2s.
Treg cells have the capacity to mediate suppression of a variety of immune cells and exert anti-inflammatory effects. In addition to the production of suppressive cytokines TGF-β and IL-10, Treg cells suppress ILC2s by inducible T cell costimulator (ICOS)–ICOS ligand interaction [57]. IL-33 induces mast cells to secrete a variety of inflammatory mediators, including IL-2. IL-2 secreted by mast cells promotes an increase in the number of IL-10–producing Treg cells and suppresses IL-33–induced airway eosinophilia, constituting an anti-inflammatory negative feedback system [58]. On the other hand, mast cell–derived prostaglandin D2 activates ILC2s via its receptor, chemoattractant receptor–homologous molecule expressed on TH2 cells (CRTH2), and mediates strong proallergic inflammatory responses [59].
DCs are a type of antigen-presenting cell. They play an essential role in promoting the adaptive immune response. ILC2-derived IL-13 promotes DC migration into draining lymph nodes, where DCs prime naïve T cells to differentiate into Th2 cells [60]. ILC2-derived IL-13 also induces the production of CCL17 by DCs, which promotes the recruitment of CCR4+ memory Th2 cells [61].
In addition, ILC2s interact with macrophages. In the context of polarized type 2 immune responses, macrophages assume a distinct state of alternative activation into M2 macrophages, which have critical functions in allergic inflammation, helminth infection, and maintenance of metabolic homeostasis. Type 2 cytokines, especially IL-13, produced by activated ILC2s promote M2 polarization, resulting in protective immunity in a cerebral malaria model and helminth infection, or induce allergic inflammation in fungal infection [62,63,64]. Moreover, M2 macrophages in visceral adipose tissue (VAT) play an important role in glucose and fat metabolism. VAT-resident ILC2s are the major source of IL-5 and IL-13; they promote the accumulation of eosinophils and M2 macrophages. These cells are required for protection from increased adiposity and insulin resistance in the context of a high-fat diet [65].
Mesenchymal cells and ILC2s
ASCs, a platelet derived growth factor receptor alpha (PDGFRα) low expressing fibroblast-like stromal cells, produces IL-33 and TSLP to support ILC2s in the peripheral microenvironment in various tissues (Fig. 5). ILC2s, in turn, promote ASC expansion and IL-33 production after helminth infection. Single-cell RNA sequencing has revealed that ASCs express high levels of collagens and cytokines and that they are associated with extracellular matrix remodeling and immune responses [30]. In adipose tissue, white adipose tissue–resident multipotent stromal cells (WAT-MSCs) act as a reservoir of IL-33 to sustain ILC2s. WAT-MSCs also support the proliferation and activation of ILC2s in an ICAM-1–LFA-1 manner.

Regulation of ILC2s by mesenchymal cells. ILC2s localize with fibroblast-like mesenchymal cells in perivascular regions. Mesenchymal cells produce IL-33 and TSLP to support ILC2s, and ILC2s promote mesenchymal cell expansion and IL-33 production during helminth infection. Mesenchymal cells not only activate ILC2s but also regulate IL-10 production via Semaphorine6D-PlexinA1 manner
A recent study revealed that lung ASCs regulate IL-10 production in ILC2s via the semaphorin 6D (Sema6D)–plexin A1 axis [66]. Semaphorins, which are axon guidance molecules, were originally identified during neuronal development. They have various effects on angiogenesis, tumor growth, bone homeostasis, and immune responses [67,68,69,70,71]. Sema6D-expressing ASCs suppress IL-10 production by ILC2s in vitro. Deletion of Sema6D ameliorates acute lung inflammation caused by IL-33 and Alternaria alternata.
Conclusions
Progress in the past several years has clarified that ILC2s are highly dynamic cells that can migrate from tissue to tissue and adapt their effector functions depending on the environment in which they reside. As mentioned earlier, in contrast to T cells and B cells, ILC2s lack antigen-specific receptors. Instead, ILC2s are directly regulated by various mediators including cytokines, lipid mediators, neurotransmitters, and cell–cell contact. Moreover, as ILC2s are tissue-resident cells, such mediators in the local microenvironment have a significant effect on the function, localization, and phenotype of ILC2s. In this review, we summarized the current knowledge regarding the mechanisms that regulate ILC2s from the perspective of the cells in the surrounding environment.
Understanding of how ILC2s are regulated highlights the therapeutic importance of targeting the surrounding mediators. For example, along with Th2 cells, ILC2s are a well-known component of an immune cell network that contributes to the pathological state of asthma. Biologics targeting IgE, IL-4, IL-5, IL-13, and TSLP are used for uncontrolled severe asthma; their targets are all mediators produced by cells in the microenvironment of ILC2s. Blood eosinophil count, fractional exhaled nitric oxide (FeNO), and blood IgE titer are used as diagnostic biomarkers for severe asthma [72,73,74]. Among these biomarkers, blood eosinophil count and FeNO are reported to be useful for predicting the efficacy of biologics in patients with severe asthma [75,76,77,78]. ILC2s produce IL-5 and enhance the proliferation, survival, and recruitment of eosinophils, which can be monitored by blood eosinophil count. However, human blood ILC2 count it is usually not measured clinically. A basic science study has shown that patients with asthma have more blood ILC2s which produce more IL-5 and IL-13 than healthy controls [79], indicating that the evaluation of blood ILC2 in patients with asthma might be clinically useful. Clinical application of ILC2 measurement in humans is a task for the future. In addition to surrounding mediators of ILC2s, regulatory functions of ILC2 might be the key to treating allergic diseases. It has been reported that allergen-specific immunotherapy restores the ability of ILC2s to produce IL-10, and IL-10–producing ILC2s play a critical role in the induction of tolerance to aeroallergens [80]. Mesenchymal cells, which regulate IL-10 production by ILC2s, might be a potential treatment target in allergic diseases.
The location of ILC2s differs between humans and mice. Moreover, ILC2s have different cytokine production profiles, receptor expression, and signaling pathways among species. Although mouse studies and human studies complement each other, we have to carefully interpret these differences.
ILC2s have been identified to be involved in many diseases related to excessive activation or dysfunction of tissue-repairing and maintaining tissue-homeostatic balance. Further clarification of the mechanisms by which ILC2s regulate the immune system will certainly shed light on the development of novel therapeutic approaches for these diseases.
Availability of data and materials
Not applicable.
Abbreviations
- ASC:
-
Adventitial stromal cell
- CGRP:
-
Calcitonin gene–related peptide
- CRTH2:
-
Chemoattractant receptor–homologous molecule expressed on Th2 cells
- DC:
-
Dendritic cell
- FeNO:
-
Fractional exhaled nitric oxide
- GATA3:
-
GATA-binding protein 3
- GWAS:
-
Genome-wide association studies
- ICOS:
-
Inducible T cell costimulator
- ILC2:
-
Group 2 innate lymphoid cell
- NE:
-
Norepinephrine
- NMU:
-
Neuromedin U
- NMUR1:
-
NMU receptor 1
- NMUR2:
-
NMU receptor 2
- Nrp1:
-
Neuropilin-1
- PNEC:
-
Pulmonary neuroendocrine cell
- Sema6D:
-
Semaphorin 6D
- S1p:
-
Sphingosine-1-phosphate
- TGFβ1:
-
Transforming growth factor beta-1
- Th2:
-
CD4+ T helper type2 cell
- Treg :
-
Regulatory T cell
- TSLP:
-
Thymic stromal lymphopoietin
- VAT:
-
Visceral adipose tissue
- VIP:
-
Vasoactive intestinal polypeptide
- WAT-MSC:
-
White adipose tissue–resident multipotent stromal cell
- α-CGRP:
-
Alpha-calcitonin gene–related peptide
- β2AR:
-
β2-Adrenergic receptor
References
Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37(4):634–48.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4.
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70.
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107(25):11489–94.
Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential “inflammatory” type 2 innate lymphoid cells. Nat Immunol. 2015;16(2):161–9.
Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. 2017;23(8):938–44.
Seehus CR, Kadavallore A, Torre B, Yeckes AR, Wang Y, Tang J, et al. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat Commun. 2017;8(1):1900.
Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA. 2015;112(34):10762–7.
Sasaki T, Moro K, Kubota T, Kubota N, Kato T, Ohno H, et al. Innate Lymphoid Cells in the Induction of Obesity. Cell Rep. 2019;28(1):202-17.e7.
Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature. 2020;579(7797):130–5.
Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62.
Gadani SP, Smirnov I, Wiltbank AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med. 2017;214(2):285–96.
Björklund ÅK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol. 2016;17(4):451–60.
Bracamonte-Baran W, Chen G, Hou X, Talor MV, Choi HS, Davogustto G, et al. Non-cytotoxic cardiac innate lymphoid cells are a resident and quiescent type 2-commited population. Front Immunol. 2019;10:634.
McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39(2):357–71.
Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R, et al. Bacteria-induced group 2 innate lymphoid cells in the stomach provide immune protection through induction of Iga. Immunity. 2020;52(4):635-49.e4.
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–50.
Popescu DM, Botting RA, Stephenson E, Green K, Webb S, Jardine L, et al. Decoding human fetal liver haematopoiesis. Nature. 2019;574(7778):365–71.
Simic M, Manosalva I, Spinelli L, Gentek R, Shayan RR, Siret C, et al. Distinct Waves from the Hemogenic Endothelium Give Rise to Layered Lymphoid Tissue Inducer Cell Ontogeny. Cell Rep. 2020;32(6):108004.
Bando JK, Liang HE, Locksley RM. Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat Immunol. 2015;16(2):153–60.
Schneider C, Lee J, Koga S, Ricardo-Gonzalez RR, Nussbaum JC, Smith LK, et al. Tissue-resident group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity. 2019;50(6):1425-38.e5.
Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol. 2018;19(10):1093–9.
Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (New York, NY). 2015;350(6263):981–5.
Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science (New York, NY). 2018;359(6371):114–9.
Ricardo-Gonzalez RR, Schneider C, Liao C, Lee J, Liang HE, Locksley RM. Tissue-specific pathways extrude activated ILC2s to disseminate type 2 immunity. J Exp Med. 2020;217(4):e20191172.
Zeis P, Lian M, Fan X, Herman JS, Hernandez DC, Gentek R, et al. In Situ Maturation and tissue adaptation of type 2 innate lymphoid cell progenitors. Immunity. 2020;53(4):775-92.e9.
Zhang J, Qiu J, Zhou W, Cao J, Hu X, Mi W, et al. Neuropilin-1 mediates lung tissue-specific control of ILC2 function in type 2 immunity. Nat Immunol. 2022;23(2):237–50.
Scott IC, Majithiya JB, Sanden C, Thornton P, Sanders PN, Moore T, et al. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep. 2018;8(1):3363.
von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. 2016;529(7585):221–5.
Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity. 2019;50(3):707-22.e6.
Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science. 2018;360(6393):eaan8546.
Xu H, Ding J, Porter CBM, Wallrapp A, Tabaka M, Ma S, et al. Transcriptional atlas of intestinal immune cells reveals that neuropeptide α-CGRP modulates group 2 innate lymphoid cell responses. Immunity. 2019;51(4):696-708.e9.
Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–90.
Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242(1):10–30.
Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. 2018;19(4):375–85.
Préfontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125(3):752–4.
Shikotra A, Choy DF, Ohri CM, Doran E, Butler C, Hargadon B, et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J Allergy Clin Immunol. 2012;129(1):104–11 e1-9.
Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J immunol (Baltimore, Md : 1950). 2005;174(12):8183–90.
Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat Commun. 2013;4:2675.
Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–46.
Corrigan CJ, Wang W, Meng Q, Fang C, Eid G, Caballero MR, et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J Allergy Clin Immunol. 2011;128(1):116–24.
Talbot S, Abdulnour RE, Burkett PR, Lee S, Cronin SJ, Pascal MA, et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron. 2015;87(2):341–54.
Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502(7470):245–8.
Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β(2)-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science (New York, NY). 2018;359(6379):1056–61.
Howard AD, Wang R, Pong SS, Mellin TN, Strack A, Guan XM, et al. Identification of receptors for neuromedin U and its role in feeding. Nature. 2000;406(6791):70–4.
Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 2017;549(7672):351–6.
Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. 2017;549(7671):282–6.
Cardoso V, Chesné J, Ribeiro H, García-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. 2017;549(7671):277–81.
Prendergast CE, Morton MF, Figueroa KW, Wu X, Shankley NP. Species-dependent smooth muscle contraction to Neuromedin U and determination of the receptor subtypes mediating contraction using NMU1 receptor knockout mice. Br J Pharmacol. 2006;147(8):886–96.
Wallrapp A, Burkett PR, Riesenfeld SJ, Kim SJ, Christian E, Abdulnour RE, et al. Calcitonin gene-related peptide negatively regulates alarmin-driven type 2 innate lymphoid cell responses. Immunity. 2019;51(4):709-23.e6.
Nagashima H, Mahlakõiv T, Shih HY, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity. 2019;51(4):682-95.e6.
Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol (Baltimore, Md : 1950). 2014;192(5):2442–8.
Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity. 2018;48(6):1195-207.e6.
Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J Exp Med. 2017;214(9):2507–21.
Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. 2014;40(5):758–71.
Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17(6):636–45.
Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol. 2017;139(5):1468-77.e2.
Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An Interleukin-33-Mast cell-interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. 2015;43(1):175–86.
Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133(4):1184–94.
Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–35.
Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol. 2016;17(1):57–64.
Besnard AG, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, et al. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog. 2015;11(2):e1004607.
Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, et al. ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun. 2015;6:6970.
Piehler D, Eschke M, Schulze B, Protschka M, Müller U, Grahnert A, et al. The IL-33 receptor (ST2) regulates early IL-13 production in fungus-induced allergic airway inflammation. Mucosal Immunol. 2016;9(4):937–49.
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–49.
Naito M, Nakanishi Y, Motomura Y, Takamatsu H, Koyama S, Nishide M, et al. Semaphorin 6D-expressing mesenchymal cells regulate IL-10 production by ILC2s in the lung. Life Sci alliance. 2022;5(11):e202201486.
Kolodkin AL, Matthes DJ, Goodman CS. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 1993;75(7):1389–99.
Pasterkamp RJ, Kolodkin AL. Semaphorin junction: making tracks toward neural connectivity. Curr Opin Neurobiol. 2003;13(1):79–89.
Toyofuku T, Zhang H, Kumanogoh A, Takegahara N, Suto F, Kamei J, et al. Dual roles of Sema6D in cardiac morphogenesis through region-specific association of its receptor, Plexin-A1, with off-track and vascular endothelial growth factor receptor type 2. Genes Dev. 2004;18(4):435–47.
Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N, Takamatsu H, et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature. 2007;446(7136):680–4.
Kang S, Kumanogoh A. Semaphorins in bone development, homeostasis, and disease. Semin Cell Dev Biol. 2013;24(3):163–71.
Ortega H, Katz L, Gunsoy N, Keene O, Yancey S. Blood eosinophil counts predict treatment response in patients with severe eosinophilic asthma. J Allergy Clin Immunol. 2015;136(3):825–6.
Wagener AH, de Nijs SB, Lutter R, Sousa AR, Weersink EJ, Bel EH, et al. External validation of blood eosinophils, FE(NO) and serum periostin as surrogates for sputum eosinophils in asthma. Thorax. 2015;70(2):115–20.
Peters MC, Kerr S, Dunican EM, Woodruff PG, Fajt ML, Levy BD, et al. Refractory airway type 2 inflammation in a large subgroup of asthmatic patients treated with inhaled corticosteroids. J Allergy Clin Immunol. 2019;143(1):104-13.e14.
Hanania NA, Wenzel S, Rosén K, Hsieh HJ, Mosesova S, Choy DF, et al. Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am J Respir Crit Care Med. 2013;187(8):804–11.
FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (London, England). 2016;388(10056):2128–41.
Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198–207.
Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N Engl J Med. 2018;378(26):2486–96.
Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134(3):671-8.e4.
Golebski K, Layhadi JA, Sahiner U, Steveling-Klein EH, Lenormand MM, Li RCY, et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity. 2021;54(2):291-307.e7.
Acknowledgements
We thank Yasutaka Motomura for a critical reading of the manuscript.
Funding
This work was supported by research grants from the Center of Innovation program (COISTREAM) from the following: the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (to A.K.); the Japan Society for the Promotion of Science (JSPS) KAKENHI (JP18H05282 to A.K.); the Japan Agency for Medical Research and Development (AMED) (J200705023, J200705710, J200705049, JP18cm016335, and JP18cm059042 to A.K.); the Kansai Economic Federation (KANKEIREN); Mitsubishi Zaidan1 (to A.K.); and the Japan Agency for Medical Research and Development–Core Research for Evolutional Science and Technology (AMED–CREST) (22gm1810003h0001 to A.K.).
Author information
Authors and Affiliations
Contributions
MN drafted the manuscript. AK revised the manuscript and gave final approval of the version to be published. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Naito, M., Kumanogoh, A. Group 2 innate lymphoid cells and their surrounding environment. Inflamm Regener 43, 21 (2023). https://doi.org/10.1186/s41232-023-00272-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-023-00272-8