
Abstract
Background
Inborn errors of metabolism are genetically inherited diseases which can lead to accumulation of toxic metabolites in the body. Inborn errors of metabolism have a high morbidity and mortality in neonates. Many inborn errors of metabolism are amenable to treatment with early diagnoses. Till now, more than 500 metabolic disorders have been detected. Although individual metabolic disorders are rare, the incidence of overall metabolic disorders is high.
Results
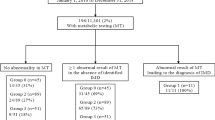
It was found that 70/200 cases (35 %) had confirmed inborn errors of metabolism, and another 8 cases (4%) suspected to have inborn errors of metabolism; 15/200 (7.5%) cases had mild elevation of phenylalanine level, while 107/200 (53.5%) had another diagnosis rather than metabolic disorders. Urea cycle defect was diagnosed in 20/70 (28.5%), maple syrup urine disease in 18/70 (25.7%), organic acidemia in 15/70 (21.4%), and non-ketotic hyperglycinemia in 1/70 (1.4 %) case. Also, 15/70 (21.4 %) cases had fatty acid oxidation defect. Lastly, one female case (1.4 %) was diagnosed to have disorder of pyrimidine deficiency.
Conclusion
Diagnosis of inborn errors of metabolism was confirmed in 35% of neonates, and 4% was suspected to have metabolic disorders. These results showed that inherited metabolic disorders are not rare. The development of a nationwide screening program for metabolic disorders is mandatory for early detection of these potentially treatable disorders.
Similar content being viewed by others

Background
Inborn errors of metabolism (IEM) are a group of inherited genetic metabolic disorders that lead to enzymatic defects in the human metabolism and form a large class of genetic diseases. The cause of IEM is the mutation in a gene that encodes for an enzyme, leading to defective synthesis of this enzyme or its activity. This affects the normal function of a metabolic pathway. In most disorders, problems arise from accumulation of substrates which are toxic to tissues, organs, or blood [1].
Till now, more than 500 IEMs have been detected. Although individual IEM is rare, the incidence of overall IEMs is high and varies dramatically in different countries and regions [2]. According to the Society for the Study of Inborn Errors of Metabolism (SSIEM) [1] and based on the main substrate, which is affected, IEM was classified as follows:
Disorders of amino acid (e.g., maple syrup urine disease, organic acidemias, and urea cycle defects)
Disorders of carbohydrate metabolism (e.g., galactosemia and glycogen storage disorders)
Disorder of lipid metabolism (e.g., fatty acid oxidation defects [medium chain acyl-CoA dehydrogenase deficiency])
Disorders in the metabolism of purines and pyrimidines
Many IEMs are amenable to treatment with early diagnoses. For others that are not amenable to treatment, establishing diagnosis in the index case is important for prenatal diagnosis in subsequent pregnancy [3].
Early diagnosis and treatment are very important to reduce the rates of morbidity and mortality related to IEMs. The first step is suspicion, followed by performing routine and specific laboratory analysis as soon as possible. Early neonatal screening is important especially in countries with high rates of consanguinity because early diagnosis and treatment are likely to result in better neurodevelopmental outcomes and lower mortality rates.
The study aimed for:
Early detection, diagnosis, and intervention to improve the neonatal outcome
Emphasizing the importance of expanded early neonatal screening
Methods
The study included 200 cases out of 5500 neonates admitted to Neonatal Intensive Care Unit. The study was conducted in the period from January 2015 to June 2018.
Inclusion criteria
-
Positive family history with affected siblings or unexplained infant/neonatal death
-
Persistent unexplained vomiting
-
Persistent metabolic acidemia
-
Convulsions
-
Hypoglycemia
-
Respiratory distress or apnea
-
Drowsiness or disturbed consciousness level
-
Poor oral intake and dehydration
Exclusion criteria
-
Multiple congenital anomalies
-
Birth anoxia
-
Birth trauma
-
Neonates with intracranial hemorrhage
-
Neonates with surgical problem
-
Neonatal sepsis based on clinical and laboratory finding
All the studied cases were subjected to (1) history taking provided by the care-giver: assessment of the following factors—sex, birth weight, gestational age, and consanguinity. The suggestive complaints of IEM were poor suckling, weak crying, convulsions, persistent vomiting, lethargy, previous sibling deaths, persistent metabolic acidosis, hypoglycemia, and/or hyperammonemia. (2) Full clinical examination: general, abdominal, cardiac, chest, and neurological examination. (3) Laboratory investigations include routine investigations (complete blood picture, C-reactive protein (CRP), liver function tests, renal function tests, serum ammonia, serum lactate, arterial blood gas, and serum glucose level), and specific metabolic investigation includes metabolic screen in blood and organic acid in urine.
Mass spectrometry analysis
Whole blood samples were drawn by heel prick or venipuncture from high-risk babies and spotted on Whatman 903 filter paper (Whatman Inc., USA). Amino acids and acylcarnitines were analyzed using a triple-quadrupole mass spectrometer, equipped with a (Acqulity, Water, USA) Waters Alliance 2790 HPLC pump.
The result of MS/MS includes levels of tyrosine, phenylalanine, citrulline, alanine, methionine, proline, leucine/isoleucine, arginine, valine, glycine, glutamic acid, aspartic acid, and argininosuccinic acid, and 29 acylcarnitines—C0, C2, C3, C3-DC, C4, C4-DC, C4-OH, C5, C5:1, C5-DC, C5-OH, C6, C6-DC, C6-OH, C8, C8-OH, C10, C10:1, C12, C12:1, C14, C14:1, C14:2, C16, C16-OH, C18, C18:1, C18-OH, and C18:1-OH—were quantified. Multiple analyte ratios were also calculated as Phe/Tyr, Leu/Phe, C3/C2, and C3-DC/C10. The internal standards were purchased from Cambridge Isotope Laboratories, and the Neolynx software (Neolynx Inc., CA, USA) was used for processing MS/MS data.
Organic acids in urine
Random urine samples were taken (5 ml) by using urine collecting bag and stored at – 20 °C for organic acid analysis by gas chromatography/mass spectrometry (GC/MS).
Test principle of organic acids in urine
Determination of organic acids in urine was done by GC/MS technique. Urine organic acid profile is an important second-line investigation in which up to 150 different disorders can be identified from a single analysis. However, some inborn errors of metabolism are clearly identifiable only during episodes of acute decompensation, e.g., medium chain acyl-CoA dehydrogenase deficiency.
Gas chromatography/mass spectrometry (GC/MS) is the most reliable method for urine organic acid analysis to detect organic acidemia and other metabolic disorders.
Statistical analysis
Data were verified, coded by the researcher, and analyzed using IBM-SPSS version 19 (@IBM-SPSS Inc., 2012, NY, USA). Data were presented as number and percentage or mean ± standard deviations. Chi-square test analysis was used to compare the proportions between qualitative variables. p value was considered statistically significant when p ≤ 0.05.
Results
The current study recruited 5500 neonates admitted to Neonatal Intensive Care Unit, in the period from January 2015 till June 2018. Highly suspected 200 cases with IEM were studied to be diagnosed.
The socio-demographic characteristics of the studied cases were 60% males (n = 122) and 78 (39%) females. Their age ranged from 1 to 30 days after birth (Table 1).
The most common manifestations of the studied cases were poor suckling (61%), followed by respiratory distress (57%), then convulsions (54%) and vomiting (52%) (Tables 2, 3, 4, 5, 6, 7, and 8).
The outcome of the cases with IEM shows that 36% of them were discharged on treatment and continue follow-up at genetic unit and the rest of the cases (64%) died due to complications.
Discussion
In the current study, consanguinity was positive in 122 patients (61%) of patients. It was higher than that reported in a study conducted by Shawky et al. [5] at Ain Shams University, Egypt, in which 17 patients (43.5%) had consanguineous parents. Also, our results were higher than a study carried out in Iraq by Arif et al. [6], in which 174 (9.9%) neonates had positive consanguineous marriage and 132 (7.5%) of their patients had positive family history of either metabolic disorders or sudden unexplained death of siblings. In this study, the presentation day ranged from 3 to 12 days with a mean of 6.5 ± 0.7 days after-birth. This was consistent with Shawky et al. [5] who reported that all patients were normal at birth with no complications during delivery, and then they started to develop symptoms at an age ranged from 1 to 10 days with a mean of 3.4 ± 2.2 days.
In our study, the most common manifestation at the time of presentation was sepsis-like manifestations (poor suckling, found in 122 out of 200 cases (61%), followed by respiratory distress in 114 out of 200 cases (57%), then convulsions in 108 out of 200 cases (54%), and vomiting in 104 out of 200 cases (52%)). Our results were in agreement with Shawky et al. [5] and Clarke [8] who reported that sepsis-like symptoms were the most common presentation (26.5% and 15%), respectively. Also, Lunda et al., 2002, reported that the predominant symptoms in neonates suggesting the possibilty of metabolic disorders were convulsions and lethargy [9].
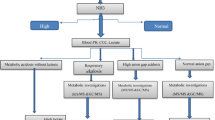
The prevalence of IEM in the current study was similar with Shawky et al. [5] and higher than that found in a study carried out by Selim et al. [10], an Iranian study by Pishva et al. [11], two studies by Elsobky and Elsayed [12] and Riyami et al. [13], an Indian one by Nagaraja et al. [14], an Iraqi one by Arif et al. [6], and two Chinese studies by Sun et al. [15] and Huang et al. [16] (Fig. 1).

Diagnostic prevalence of the studied cases
Shawky et al. [5] showed that 13/40 (35.5%) suspected neonates were admitted to the El Mahalla Hospital NICU, Egypt, over 1-year duration with sepsis-like symptoms, convulsions, persistent metabolic acidosis, persistent vomiting, and previous sibling death of unidentified causes confirmed to have IEM.
Moreover, Pishva et al. [11] reported that the prevalence of IEM was 14%, which was lower than in our study (Fig. 2). Another study in Karachi by Wajner et al. [17] reported the prevalence of 25.8%. Moreover, several other studies, in Egypt by Elsobky and Elsayed [12], in Brazil by Coelho et al. [18], and in India by Nagaraja et al. [14], reported lower detection rate 6.5%, 3.17%, and 3.2%, respectively.

IEM Subcategory prevalence among the studied IEM cases
Additionally, a study done in Oman by Riyami et al. [13] found that 10.8% of their patients were diagnosed with IEMs in high-risk Omani neonates. Likewise, an Iraqi study by Arif et al. [6] with a total number of 1758 symptomatic Iraqi children from birth to early adolescence was analyzed for possible IEM; positive cases were 224/1758, with a rate of detection (12.7%) both of which were lower than in our study.
Amino acid disorder (AA) in our study was accounted as the most common disorder detected, and this was in accordance with Selim et al. [10]. Regarding the AA disorders, urea cycle disorders (UCD) was the most common type in our study, detected in 20/70 (28.5%) of diagnosed cases, with male predominance in 15/20 case (75%). This was in agreement with Shawky et al. [5] as UCD was the most common type with an incidence but with higher level than our study that was 53.8%, and 75% of the diagnosed cases were males. In Shawky et al.’s study [5], the most common IEM was UCD in 7/13, then MSUD in 2/13, followed by MMA and MCAD 1/13. Dissimilar, Gündüz et al. [4] found that UCD was detected only in six cases (11.8%), which were lower than in our study for both.
The second most common amino acid disease in our study was maple syrup urine disease (MSUD), which was detected in 18 cases (25.7%). Our results were higher than in Shawky et al. [5] who diagnose 2/13 (15.4%) cases with MSUD and Selim et al. [10] which was 7.4%. Similarly, Karam et al. [19] in Lebanon identified MSUD in 9.1%; in Fateen et al. [20] in Egypt, it was 1.2%; and Elsobky and Elsayed [12] reported that 10% of children with convulsions had MSUD. This can be explained by the larger number of patients in our study complaining from poor suckling and convulsions, and these presentations are considered as the common symptoms of MSUD.
Furthermore, in our study, organic acidemia was presented in 15/70cases (21.4%), which was higher than in Shawky et al. [5] who detect organic acidemia in 3/40 (7.5%) and in Elsobky and Elsayed [12] who reported organic acidemia in 1.07%.
In our study, one case was diagnosed as B-ketothiolase deficiency; this case was presented on the 1st day after birth with persistent vomiting, convulsion, metabolic acidosis, and hypoglycemia. This patient had one sister with the same disease diagnosed after birth with increased tiglylglycine in urine. In our study, fatty acid oxidation defect was presented in 15/70 cases (21.4 %). This was higher than Riyami et al. [13] in Oman, who detect FAOD in (16%) of cases; Shawky et al. [5] who found FAOD in 1/13 (7.6%); in Selim et al. [10] who found FAOD in 3.4% of patients; and in Arif et al.’s study [6] in Iraq (1.1%).
Our results were lower than that of Vargas et al. [7] who reported that FAOD was detected in 19 patients (28%). These higher results in their study can be explained by their selection criteria which were based on selection of patients with FAOD manifestations only.
In our study, galactosemia was suspected in 3/200 (1.5%) cases. Our results were lower than a study by Fateen et al. [20], who detected galactosemia in (2.5%) of cases. As well, a study by Shawky et al. [5] detected galactosemia in (7.7%) and in a study by Arif et al. [6], who detected galactosemia in 31/1758 (1.76%) of cases. This difference might be explained by that more neonates in their studies presented with clinically suspicious galactosemia and have facilities to do enzymatic assay for galactosemia that was not available in our hospital. Also, they included larger number of patients for longer time.
Conclusion
From this descriptive study, we concluded that IEM disorders are not rare disease in high-risk neonates with attentions to consanguinity which is a common tradition in our country. Regarding the metabolic disorders, manifestation presented in most cases, i.e., sepsis-like manifestations. These manifestations are common, and many physicians misdiagnose them as they are unaware about these disorders. IEMs represented a significant cause of sick neonate’s admission to NICU, and it should be considered as a differential diagnosis for any sick neonate. Most IEMs are inherited in an autosomal recessive pattern, so its incidence increases with positive consanguinity. The delay in diagnosis of IEM (due to lack of facilities) leads to high morbidity and mortality, so early detection and early intervention to the neonates at risk of IEMs before the onset of symptoms can prevent or, at least, reduce serious neurological and developmental squeal.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and are available from the corresponding author on reasonable request.
References
Ezgu F. Inborn errors of metabolism. Advances in clinical chemistry. 73: Elsevier; 2016. p. 195-250.
Campeau PM, Scriver CR, Mitchell JJ (2008) A 25-year longitudinal analysis of treatment efficacy in inborn errors of metabolism. Molecular genetics and metabolism. 95(1-2):11–16
Cloherty JP, Eichenwald EC, Stark AR (2008) Manual of neonatal care: Lippincott Williams & Wilkins
Gündüz M, Ünal S, Okur İ, Ayranci-Sucakli I, Güzel F, Koç N (2015) Neonates with inborn errors of metabolism: spectrum and short-term outcomes at a tertiary care hospital. The Turkish journal of pediatrics. 57(1):45
Shawky RM, Abd-El Khalek HS, Elakhdar SE (2015) Selective screening in neonates suspected to have inborn errors of metabolism. Egyptian Journal of Medical Human Genetics. 16(2):165–171
Arif A, Thejeal RF, Farhan A (2016) Inborn errors of metabolism status in Iraq. IOSR J Pharm Biol Sci. 11:58–62
Vargas CR, Ribas GS, da Silva JM, Sitta A, Deon M, de Moura CD et al (2018) Selective Screening of fatty acids oxidation defects and organic acidemias by liquid chromatography/tandem mass spectrometry acylcarnitine analysis in Brazilian patients. Archives of medical research. 49(3):205–212
Clarke S (2002) Tandem mass spectrometry: the tool of choice for diagnosing inborn errors of metabolism? British journal of biomedical science. 59(1):42–46
Lund A, Christensen E, Skovby F (2002) Diagnosis and acute treatment of inborn metabolic diseases in infants. Ugeskrift for Laeger. 164(48):5613–5619
Selim LA, Hassan SA-H, Salem F, Orabi A, Hassan FA, El-Mougy F et al (2014) Selective screening for inborn errors of metabolism by tandem mass spectrometry in Egyptian children: a 5 year report. Clinical biochemistry. 47(9):823–828
Pishva N, Mirzaee A, Karamizade Z, Pourarian S, Hemmati F, Razvi M et al (2015) Selective screening of 650 high risk Iranian patients for detection of inborn error of metabolism. Iranian Journal of Neonatology IJN. 5(4):11–14
Elsobky E, Elsayed SM (2004) Extended metabolic screen in sick neonates and children. Egypt J Med Hum Genet. 5(2):1–7
Al Riyami S, Al Maney M, Joshi SN, Bayoumi R (2012) Detection of inborn errors of metabolism using tandem mass spectrometry among high-risk Omani patients. Oman medical journal. 27(6):482
Nagaraja D, Mamatha SN, De T, Christopher R (2010) Screening for inborn errors of metabolism using automated electrospray tandem mass spectrometry: study in high-risk Indian population. Clinical biochemistry. 43(6):581–588
Sun W, Wang Y, Yang Y, Wang J, Cao Y, Luo F et al (2011) The screening of inborn errors of metabolism in sick Chinese infants by tandem mass spectrometry and gas chromatography/mass spectrometry. Clinica Chimica Acta. 412(13-14):1270–1274
Huang X, Yang L, Tong F, Yang R, Zhao Z (2012) Screening for inborn errors of metabolism in high-risk children: a 3-year pilot study in Zhejiang Province. China. BMC pediatrics. 12(1):18
Wajner M, de Moura CD, Ingrassia R, de Oliveira AB, Busanello ENB, Raymond K et al (2009) Selective screening for organic acidemias by urine organic acid GC–MS analysis in Brazil: Fifteen-year experience. Clinica Chimica Acta. 400(1-2):77–81
Coelho JC, Wajner M, Burin MG, Vargas C, Giugliani R (1997) Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. European journal of pediatrics. 156(8):650–654
Karam PE, Habbal M-Z, Mikati MA, Zaatari GE, Cortas NK, Daher RT (2013) Diagnostic challenges of aminoacidopathies and organic acidemias in a developing country: a twelve-year experience. Clinical biochemistry. 46(18):1787–1792
Fateen EM, Ibrahim M, Abdallah Z (2014) Fifteen years experience: Egyptian metabolic lab. Egyptian Journal of Medical Human Genetics. 15(4):379–385
Acknowledgements
We acknowledge the medical staff of Neonatal Intensive Care Unit, Assiut University Children Hospital, and staff member of Genetics Unit at Assiut Children Hospital and Ain Shams Genetic Unit. It was not possible to complete this work without the help and approval of the neonates’ caregivers.
Funding
This study was funded by Faculty of Medicine, Assiut University, for detection of metabolic disorders in neonates admitted at Assiut University Children Hospital.
Author information
Authors and Affiliations
Contributions
SMK was responsible for the conception and design of the study, acquisition of data, analysis and interpretation of data, critical revision of the submitted protocol for important intellectual content, statistical analysis, and supervision. MME was responsible for the conception and design of the study, analysis and interpretation of data, critical revision of the submitted protocol for important intellectual content, statistical analysis, and supervision. NHR was responsible for the conception and design of the study, analysis and interpretation of data, critical revision of the submitted protocol for important intellectual content, statistical analysis, and supervision. AMA was responsible for the conception and design of the study, analysis and interpretation of data, technical, material support, and supervision. All authors had read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the corresponding Ethical Review Board at Assiut University (N: 1050, 14/07/2015), and written informed consent was taken from patients’ parents.
The work has been performed in accordance with Helsinki Declaration consent for publication.
The authors agree for the publication of identifying images or other personal or clinical details of the participants.
Consent for publication
Consent for publication data was taken from patients’ parents, and they agree for the publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Khalaf, S.M., El-Tellawy, M.M., Refat, N.H. et al. Detection of some metabolic disorders in suspected neonates admitted at Assiut University Children Hospital. Egypt J Med Hum Genet 20, 29 (2019). https://doi.org/10.1186/s43042-019-0030-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-019-0030-5