
Abstract
Background
The purpose of this study was to characterize subtype-specific patterns of mRNA and miRNA expression of gliomas using The Cancer Genome Atlas (TCGA) data to search for genetic determinants that predict prognosis in terms of overall survival and to create interaction networks for grade 2 and 3 (G2 and G3) astrocytomas, oligodendrogliomas and grade 4 (G4) glioblastoma multiforme. Based on open-access TCGA data, 5 groups were formed: astrocytoma G2 (n = 58), astrocytoma G3 (n = 128), oligodendroglioma G2 (n = 102), oligodendroglioma G3 (n = 72) and glioblastoma G4 (n = 564); normal samples of brain tissue were also analysed (n = 15). Data of patient age, sex, survival and expression patterns of mRNA and miRNA were extracted for each sample. After stratification of the data into groups, a differential analysis of expression was carried out, genes and miRNAs that affect overall survival were identified and gene set enrichment analysis (GSEA) and interaction analysis were performed.
Results
A total of 939 samples of glial tumours were analysed, for which subtype-specific expression profiles of genes and miRNAs were identified and networks of mRNA-miRNA interactions were constructed. Genes whose aberrant expression level was associated with survival were determined, and pairwise correlations between differential gene expression (DEG) and differential miRNA expression (DE miRNA) were calculated.
Conclusions
The developed panel of genes and miRNAs allowed us to differentiate glioma subtypes and evaluate prognosis in terms of the overall survival of patients. The regulatory miRNA-mRNA pairs unique to the five glioma subtypes identified in this study can stimulate the development of new therapeutic approaches based on subtype-specific mechanisms of oncogenesis.
Similar content being viewed by others


Background
Gliomas are the most common type of tumours of the central nervous system and are characterized by a very poor prognosis, mainly due to the complexity of the treatment and the lack of targets suitable for developing effective therapeutic approaches. However, recent progress in molecular studies of gliomas is reflected in their classification and potential treatment goals. The main criteria for the modern classification of glial tumours (2016 edition of the World Health Organization classification) are the degree of malignancy (grades 1–4) and molecular markers [1, 2]. However, the heterogeneity of gliomas is the main obstacle to their accurate molecular classification, and therefore, single markers are not enough to subtype and select the optimal treatment regimen.
With the development of next-generation sequencing technologies, researchers are now able to determine the most detailed molecular genetic profile for each sample. This influx of big data generated a need to create such large consortia as The Cancer Genome Atlas (TCGA), which allow accumulation of huge amounts of data for collaborative research of various types of neoplasia. Such studies have led to the construction of interactive mRNA-miRNA networks demonstrating regulatory mechanisms that are specific to certain nosologies [3,4,5,6]. The study of the expression profiles of mRNA and miRNA of various subtypes of gliomas and the construction of RNA-miRNA interaction networks can serve as a prerequisite for the development of highly accurate diagnostic tests and the development of personalized therapeutic approaches that will increase the life expectancy and improve the patient's physical condition.
The aims of our study were to research subtype-specific patterns of mRNA and miRNA expression of diffuse gliomas using TCGA data, search for genetic determinants that determine the prognosis in terms of overall survival and construct interaction networks for glial tumours of various grades.
Methods
Sample and data collection
Based on open-access TCGA data, 5 main groups of samples were formed: diffuse astrocytoma grade 2 (G2), anaplastic astrocytoma G3, diffuse oligodendroglioma G2, anaplastic oligodendroglioma G3 and glioblastoma (GBM), and these groups of samples were subjected to RNA and miRNA sequencing analysis (TCGA-GBM and TCGA-LGG datasets), as well as microarray analysis of miRNA data (for GBM) (Fig. 1). Normal brain tissues (n = 15) were used as comparison samples. Data collection and processing were performed using R 3.6.2 software (“TCGABiolinks” [7] and “GDCRNATools” packages [8]). The miRNA names were updated to miRBase version 21 using the “miRBaseConverter” package [9].

Scheme for the processing and analysis of data on gene and microRNA expression in samples of glial tumours
The clinical and demographic characteristics of the patients are shown in Table 1.
Analysis of differential gene and miRNA expression
For the differentially expressed gene (DEG) and differentially expressed microRNA (DE miRNA) analyses, the “limma” package [10] and the glmLRT test (generalized linear models likelihood ratio test) were used. The cut-off points were FDR < 0.01 and | logFC | > 1. The results are presented in the form of a scatter plot (volcano plot).
Gene set enrichment analysis
The genes selected in the analysis of differential expression were divided into groups depending on their genetic ontology (molecular functions, biological processes, cell localization and signalling pathways) using the “TCGAbiolinks” package [7] and the DAVID (Database for Annotation, Visualization and Integrated Discovery) database [11].
Survival analysis
The survival analysis involved several steps. First, genes were selected using the Kaplan–Mayer method in the TCGABiolinks package, the expression of which had a significant effect on the duration of overall survival. The expression of each gene with the quantile cut-off points (upper—0.6 and lower—0.33) was divided into low, moderate and high. The effect of the genes with high and low expression on survival was then evaluated using Cox multivariate regression analysis. Genes selected in the regression analysis were clustered based on the protein-protein interactions of their products (via the STRING database) [12]. We visualized the dependence of overall survival on the expression of the genes selected on the basis of the previous two analyses and interactive analysis using the “UCSCXenaTools” [13], “survival” and “survminer” packages.
Interactome analysis
Correlation analysis of gene and miRNA expression was performed for screening (via the “Hmisc” package). Gene-miRNA pairs with a Pearson correlation coefficient greater than − 0.5 were excluded. The p value threshold used for statistical significance was equal to 0.05. The remaining pairs were verified using ten databases (“multiMiR” package) [14]: miRecords, miRTarBase, TarBase, DIANA-microT-CDS, ElMMo, MicroCosm, miRanda, miRDB, PicTar and PITA. The final network of miRNA-target interactions included validated pairs (databases showed a status of “validated”). Predicted pairs (databases showed a status of “predicted”) were selected based on the overall survival result. The interaction network was built using the “igraph” package.
Results
Analysis of differential gene expression
A total of 939 glial tumour samples were analysed. Of the studied glioma subtypes, the DEGs (Table 2) were especially obvious for diffuse astrocytoma (Fig. 2).

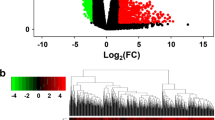
Volcano plot of differentially expressed genes in diffuse astrocytoma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (p value with FDR correction). Genes with logFC > 1 and logFC < − 1 (with a p value < 0.05) are marked in red and green, respectively. The labels indicate the “top 10” genes with reduced/increased expression. (Figures for the remaining subtypes are given in the Supplementary material: Online Resource 1 for anaplastic astrocytoma, Online Resource 2 for diffuse oligodendroglioma, Online Resource 3 for anaplastic oligodendroglioma, and Online Resource 4 for glioblastoma.)
When compared with non-tumour samples, diffuse astrocytoma samples showed 3392 DEGs, and anaplastic astrocytomas showed 3387 DEGs. The aberrant tumour expression profile was dominated by cases of increased transcriptional activity in astrocytoma G2 samples (1988 upregulated vs. 1404 downregulated) and, conversely, decreased expression in astrocytoma G3 samples (1400 upregulated vs. 1987 downregulated). In the GBM group, 2324 genes had differentially increased expression, and 2491 genes had differentially reduced expression. A slightly smaller number of differentially expressed genes was described for oligodendrogliomas (Table 2): 2723 DEGs in diffuse oligodendroglioma samples and 2896 DEGs in anaplastic oligodendroglioma samples compared with non-tumour samples. For two subtypes of glial tumours, DEGs with reduced expression prevailed: 1672 upregulated vs. 1051 downregulated genes in oligodendroglioma G2 samples and 1738 upregulated vs. 1158 downregulated genes in oligodendroglioma G3 samples.
When assessing the genetic ontology of the DEGs of astrocytomas, genes associated with EIF2 signalling, axonal guidance and neuropathic pain signalling in dorsal horn neurons were generally identified (Table 3). In addition, DEGs associated with G protein-coupled receptors and cAMP-mediated signalling were determined for diffuse astrocytomas, and role the DEGs of diffuse anaplastic astrocytomas were enriched in processes related to NFAT in cardiac hypertrophy and synaptic long-term potentiation.
Gene ontology analysis (Table 3) made it possible to identify common significant signalling pathways for oligodendrogliomas, such as the EIF2 pathway, mTOR signalling and dopamine–DARPP32 feedback in cAMP signalling, as well as subtype-specific pathways. The top signalling pathways of diffuse oligodendroglioma samples included axonal guidance signalling and neuropathic pain signalling in dorsal horn neurons, while the top pathways included G protein-coupled receptor signalling and synaptic long-term potentiation for anaplastic oligodendroglioma samples. It is worth noting that expression related to synaptic long-term potentiation signalling was aberrant in both astrocytoma and oligodendroglioma G3 samples, suggesting that it may be related to advanced stage (Table 3).
The top five signalling pathways for GBM samples were axonal guidance signalling, Rho family GTPases pathway, breast cancer regulation by stathmin1, molecular mechanisms of cancer and IL-8 signalling. In this case, the last four of the listed pathways were able to differentiate GBM into a separate group.
Figure 3 illustrates the distribution of genes that are differentially expressed either in all the tumours analysed, in some subtypes or a specific subtype. As a result, 1036 upregulated and 517 downregulated universal DEGs were revealed in samples of glial tumours. Of the 1553 nonspecific DEGs, the most represented were participants in the following signalling pathways: calcium, MAPK, oxytocin and neuroactive ligand receptor pathways. These signalling pathways can mediate neural network activity and cell proliferation. A more pronounced overlap of aberrant DEGs was observed between the GBM, astrocytoma G2 and astrocytoma G3 groups, which can be explained by the astrocytic origin of GBM.

Distribution of DEGs across 5 subtypes of glial tumours. a DEGs with logFC < − 1. b DEGs with logFC > 1
Gene ontology analysis of the 210 unique DEGs in diffuse astrocytomas, shown as an example in Fig. 4, identified changes in transcriptional activity of genes encoding passive transmembrane transport proteins, which are located mainly on the plasma membrane of cells (Fig. 4).

Genetic ontology of DEGs specific to diffuse astrocytoma. a Biological process. b Cellular component. c Molecular function. d Pathways. (Figures for the remaining subtypes are given in the Supplementary material: Online Resource 5 for anaplastic astrocytoma, Online Resource 6 for diffuse oligodendroglioma, Online Resource 7 for anaplastic oligodendroglioma, and Online Resource 8 for glioblastoma.)
The 119 DEGs specific to astrocytoma G2 samples were identified to be involved in the biological processes of translation and translational elongation. Most of the DEGs perform the molecular function of transmembrane transport or are structural components of ribosomes. Sixty-three DEGs specific to astrocytoma G3 samples were found to generally be involved in translational elongation and ion transport, as were the 147 DEGs specific to oligodendroglioma G2 samples (Online Resource 6). GBM was obviously different from the other subtypes, demonstrating 1716 unique DEGs, some of which were found to be involved in biological processes such as sensory perception of chemical stimuli, cognitive and systemic neurological processes and performing transmembrane transport functions.
Analysis of differential miRNA expression
The results of evaluating the differential expression of miRNAs in different types of glial tumours are shown in Table 2. As an example, the results for diffuse astrocytomas are illustrated in Fig. 5. Of note, the number of identified DE miRNAs was an order of magnitude less than the number of identified DEGs. Moreover, there were ~ 2 times more upregulated DE miRNAs than downregulated DE miRNAs for astrocytomas and oligodendrogliomas. The number of GBM-specific miRNAs was an order of magnitude less than the number of GBM-specific DEGs, and their distribution according to the degree of altered activity was approximately equal (Table 2).

Volcano plot of differential miRNA expression in diffuse astrocytoma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (p value with FDR correction). Genes with logFC > 1 and logFC < − 1 (with a p value < 0.05) are marked in red and green, respectively. The labels indicate the “top 10” miRNAs with reduced/increased expression. (Figures for the remaining subtypes are given in the Supplementary material: Online Resource 9 for anaplastic astrocytoma, Online Resource 10 for diffuse oligodendroglioma, Online Resource 11 for anaplastic oligodendroglioma, and Online Resource 12 for glioblastoma.)
Venn diagrams (Fig. 6) were constructed to show the differential expression of miRNAs depending on the glioma subtype.

Distribution of DE miRNAs across 5 subtypes of glial tumours. a DE miRNAs with logFC < − 1. b DE miRNAs with logFC > 1
As a result, 35 microRNAs were identified, and the degree of change for these microRNAs was similar for all types of glial tumours, including GBM. It should be noted that 207 common DE miRNAs were identified in astrocytoma and oligodendroglioma G2 and G3 samples, while GBM samples had only 69 DE miRNAs common to other subtype samples. The largest number of specific small noncoding RNAs was determined for G3 tumours: 65 (vs. 21) miRNAs discriminated astrocytoma G3 samples; and 51 (vs. 10) miRNAs discriminated oligodendroglioma G3 samples.
Association of overall survival with the expression of markers differentiating the studied subtypes of glial tumours
Based on the initial screening data, a pool of genes was formed, the differential expression of which not only was unique for each subtype but also correlated with the overall survival of patients (Online Resource 20). The resulting panel included 26 DEGs unique to astrocytoma G2; 15 DEGs unique to astrocytoma G3; 17 DEGs unique to oligodendroglioma G2; 7 DEGs unique to oligodendroglioma G3; and 151 DEGs unique to GBM. Table 4 summarizes the unique survival-related DEGs that were not previously mentioned in glial tumour studies in PubMed and Cochrane.
Unique DE miRNAs, whose targets were found to be associated with survival, were identified for three subtypes of glial tumours (Online Resource 21): astrocytoma G2 (hsa-let-7c-5p and hsa-miR-125b-5p); astrocytoma G3 (hsa-miR-190b, hsa-miR-196a-3p, and hsa-miR-218-2-3p); and oligodendroglioma G3 (hsa-miR-20b-5p, hsa-miR-466, and hsa-let-7a-5p).
Interactome analysis of miRNA-mRNA interactions
For each tumour subtype, an analysis of DEG associations with overall survival was performed. A total of 360 and 835 DEGs were identified in astrocytoma G2 and G3 samples, respectively, and 227 and 468 DEGs were identified in oligodendroglioma G2 and G3 samples, respectively. Genes that correlated with survival were clustered depending on the significance level of PPIs (protein-protein interactions according to STRING analysis). For example, 6 clusters were revealed for astrocytoma G2 samples (Fig. 7), 10 clusters were revealed for astrocytoma G3 samples and 1, 8 and 8 clusters were revealed for oligodendroglioma G2, oligodendroglioma G3 and GBM samples, respectively (Online Resource 13, 14, 15, 16).

Interactome analysis of genes associated with overall survival in diffuse astrocytoma clustered according to the p value of protein-protein interactions. (Figures for the remaining subtypes are given in the Supplementary material: Online Resource 13 for anaplastic astrocytoma, Online Resource 14 for diffuse oligodendroglioma, Online Resource 15 for anaplastic oligodendroglioma, and Online Resource 16 for glioblastoma.)
Thereafter, Pearson correlation analysis was performed on the expression levels of selected genes and miRNAs, and pairs with correlation values of − 0.5 or lower were considered significant. Figure 8 presents an example of the results of the interactome analysis for diffuse astrocytoma, and mRNA-miRNA pairs, which were verified against databases of validated/predicted miRNA targets, are visualized.

Scheme of the interactions of miRNAs and the mRNA of genes associated with survival in the studied samples of diffuse astrocytoma. The red colour denotes DE miRNAs, the green colour denotes DEGs. (Figures for the remaining subtypes are given in the Supplementary material: Online Resource 17 for anaplastic astrocytoma, Online Resource 18 for diffuse oligodendroglioma, and Online Resource 1 for anaplastic oligodendroglioma.)
Thus, 173 DE miRNA and DEG mRNA pairs were associated with survival in the samples of diffuse astrocytoma, 76 DE miRNA and DEG mRNA pairs were associated with survival in diffuse oligodendroglioma samples and 199 and 372 DE miRNA and DEG mRNA pairs were associated with survival in anaplastic astrocytoma and anaplastic oligodendroglioma, respectively. For GBM, no correlation was found between DEGs and DE miRNAs.
Discussion
In our study, for each of the five subtypes of glioma, specific gene and microRNA expression profiles were described (Table 2). As a result, patterns of specifically expressed markers were determined: 211 unique DEGs and 21 DE miRNAs were identified in astrocytoma G2, 63 and 65 were identified in astrocytoma G3, 119 and 10 were identified in oligodendroglioma G2 and 147 and 51 were identified in oligodendroglioma G3.
The identified expression patterns were generally related to signalling pathways associated with neuronal activity, proliferation and cancer progression. In glial tumour samples at earlier stages (G2 and G3 samples), a similarity in the set of signalling pathways with aberrant activity was demonstrated (Table 3). At the same time, signalling pathways that were not shared by the astrocytomas or oligodendrogliomas were identified for the GBM group.
Based on the initial screening data, a panel of genes was developed, the differential expression of which not only was unique for each subtype but also correlated with the overall survival of patients (Online Resource 20). The panel includes 26 DEGs unique to astrocytoma G2, 15 DEGs unique to astrocytoma G3, 17 DEGs unique to oligodendroglioma G2, 7 DEGs unique to oligodendroglioma G3 and 151 DEGs unique to GBM.
Currently, the role of individual miRNAs in glial tumours is not well understood. MiRNAs are small non-coding regulatory RNAs that decrease the stability and/or inhibit the translation of target mRNAs with which they have full or partial complementarity. These regulatory molecules, playing an important role in the pathogenesis, development and progression of cancer and the response to therapy, are especially promising for diagnostic strategies, including minimally invasive methods (measuring the level in plasma, serum or cerebrospinal fluid).
Primary data analysis via TCGA allowed us to differentiate diffuse astrocytomas and oligodendrogliomas from other subtypes of gliomas by the expression of 21 and 10 miRNAs, respectively; anaplastic astrocytoma and oligodendroglioma could be differentiated from other gliomas based on the expression of 65 and 51 miRNAs, respectively. For the GBM group, 13 unique DE miRNAs were identified. After the Pearson correlation analysis and the validation of the correlation pairs in the databases, unique miRNAs that were found to target the DEGs were identified for some subtypes, and these miRNAs were associated with survival. Below, we briefly consider their characteristics.
In diffuse astrocytomas, a decrease in the levels of hsa-let-7c-5p and hsa-miR-125b-5p was detected. In parallel, it was noted that the expression of hsa-let-7c-5p was associated with the level of transcriptional activity of the BEGAIN gene; the link between hsa-miR-125b-5p and the mRNA level of the DUSP3 and RAB3D genes was similarly found. Earlier, in an experimental study on cells of acute erythroleukaemia, the antiproliferative effect of let-7c-5p was demonstrated to be associated with the targeting of the PBX2 oncogene [24]. In colorectal cancer cells after exposure to 5-FU or starvation, Hou N et al. identified a miRNA pool including hsa-let-7c-5p [25]. The identified correlation between the expression of hsa-let-7c-5p and BEGAIN is consistent with the presence of one binding site (target score—90) within the 3′-UTR of the mRNA of this gene.
The BEGAIN protein is involved in the regulation of the activity of postsynaptic neurotransmitter receptors (GO: 0098962); the contribution of this locus to oncogenesis has not been confirmed to date. The antitumour effect of miR-125b has been described in several studies of lung cancer, hepatocellular carcinoma and breast cancer [26,27,28,29]. For example, the study of Yuan M et al. demonstrated the antiproliferative effect of increased expression of hsa-miR-125b-5p in cell lines of diffuse astrocytoma in children [30]. In contrast, a number of studies of endometrial cancer, prostate cancer and GBM indicate the oncogenic effects of hsa-miR-125b-5p, which targets the known tumour suppressors TP53 and GJA1 [31,32,33]. The target of hsa-miR-125b-5p in diffuse astrocytomas is probably the mRNA of the DUSP3 gene, a member of a subfamily of protein phosphatases that negatively regulate the members of the mitogen-activated protein kinase superfamily (MAPK/ERK, SAPK/JNK and p38) and in such a way control proliferation and differentiation. The RAB3D protein (which has three 3′-UTR binding sites for miR-125b-5p) is described as an oncogenic small GTPase that promotes both cell migration in glial tumours and metastasis in breast cancer via the intracellular AKT/GSK3β signalling pathway [34, 35]. In a single tumour, hsa-miR-125b-5p exhibits both oncosuppressive and oncogenic effects.
Anaplastic astrocytomas were characterized by a change in the level of three unique miRNAs: hsa-miR-190b-3p, hsa-miR-196a-3p and hsa-miR-218-2-3p. Correlation analysis allowed us to associate the level of hsa-miR-190b-3p with the expression of the EFNB1 gene, which is involved in cell adhesion and the formation of neural synapses. In this case, the expression of the hsa-miR-190b-3p target was increased, which can be explained by the presence of additional mechanisms regulating the transcription of EFBN1. However, hsa-miR-190b-3p may mitigate the increased expression of EFBN1. An increase in the level of hsa-miR-190b-3p has also been shown in other types of malignant neoplasms. For example, validation of an analysis of 1083 breast cancer samples (from TCGA) confirmed an increased level of expression of hsa-miR-190b-3p in both surgical biopsies and breast cancer cell lines, and this overexpression significantly contributed to cell proliferation and migration [36]. The expression of hsa-miR-190b-3p correlates with the expression of the C8orf34 gene, which is involved in the regulation of gene expression and the cell cycle. Mutations at this genetic locus can mediate severe toxicity during chemotherapy [37].
The increased activity of hsa-miR-196a-3p in colorectal cancer was demonstrated by the study of Wang YN [38]. Similarly, Chen Y. et al. showed that a decrease in hsa-miR-196a-3p expression was associated with a high risk of metastasis in breast cancer. The authors found that transforming growth factor β1 negatively regulates the expression of hsa-miR-196a-3p and activates neuropilin-2, which causes the migration and invasion of tumour cells [39].
Our study demonstrated a decrease in hsa-miR-218-2-3p expression. As a result of the correlation analysis and subsequent database validation, the correlation of hsa-miR-218-2-3p and LMO7 expression was revealed. In contrast, earlier in the Feng Z study, overexpression of hsa-miR-218-2-3p was detected both in cell lines and in surgical biopsy specimens of glial tumours of various grades. In the study, an increase in the activity of has-miR-218-2-3p was associated with the invasive and proliferative potential of glial tumours [40]. As a possible target of hsa-218-2-3p, LMO7 is described as a transcription factor and stabilizer of adhesion compounds, a change in the expression of which is associated with some oncological nosologies [41, 42]. Decreased LMO7 expression in mouse models leads to the development of lung adenocarcinoma [43], and low expression in human lung adenocarcinoma was associated with a negative prognosis [42]. However, contrasting results have also been shown, in which high expression of LMO7 was identified as a negative prognostic factor in non-small-cell lung cancer associated with LRIG1 expression [44].
Despite the numerous miRNA-mRNA correlation pairs (n = 75) identified in diffuse oligodendrogliomas, there were no subtype-specific miRNAs among them.
The analysis made it possible to distinguish three DE miRNAs unique to anaplastic oligodendrogliomas: hsa-miR-20b-5p, hsa-miR-466 and hsa-let-7a-5p.
Earlier, in a study of colorectal cancer (CRC), the association of hsa-miR-20b-5p with survival, as well as a negative correlation of this miRNA with one of the E2F5 cell cycle regulators, was noted [45]. In addition, increased levels of this microRNA in the blood were associated with a better prognosis in patients with metastatic CRC receiving chemotherapy based on bevacizumab [46]. Our analysis demonstrated a negative correlation between hsa-miR-20b-5p expression and PRKACB. This genetic locus has been identified as a significant oncogene involved in the progression of endocrine cancer by modulating the signalling activity of cAMP [47, 48]. In turn, non-small-cell lung cancer shows a lower expression level of PRKACB than normal tissues, and an increase in PRKACB transcriptional activity reduces the number of proliferative, colony-forming and invasive cells and increases the incidence of apoptosis [49].
In a study devoted to the development of predictors of GBM treatment, high expression of hsa-let-7a-5p was interpreted as a favourable prognostic factor associated with survival [50]. In breast cancer, hsa-let-7a-5p mediates sensitivity to the proteasome inhibitor bortezomib, regardless of the molecular subtype [51]. The potential significance of let-7a-5p was determined by Zhou X and colleagues in a study of non-small-cell lung cancer [52].
Using layer-by-layer data filtering, the hsa-let-7a-5p and CYP46A1 correlation pair was identified. The CYP46A1 genetic locus encodes cholesterol 24-hydroxylase, an enzyme responsible for eliminating cholesterol in 24S-hydroxycholesterol (24S-OHC, oxysterol) that is specific to the brain [53]. A decrease in CYP46A1 expression was demonstrated in a study by Han M et al., in which in silico analysis of DEGs and subsequent in vitro verification were performed for GBM samples. Single-cell transcriptome sequencing data demonstrated prevailing CYP46A1 expression in neurons, astrocytes and oligodendrocyte progenitor cells compared to tumour cells [54]. Overexpressed CYP46A1 modulated a decrease in colony formation, proliferation and generation of tumour spheroids in vitro due to reduced cholesterol accumulation [54].
The level of hsa-miR-466 was found to be decreased in osteosarcoma and correlated with IRS1 expression. Increased expression of has-miR-466 inhibited the in vitro proliferation and invasion of osteosarcoma cells [55]. Similarly, a decrease in hsa-miR-466 expression has been demonstrated in hepatocellular carcinoma cells, and an increase in hsa-miR-466 expression inhibits proliferation, induces apoptosis and reduces the metastatic potential of the tumour by targeting MTDH [56]. In prostate cancer, a decrease in hsa-miR-466 expression was demonstrated due to the activity of the long non-coding RNA TUC338. The authors also revealed the antiproliferative potential of hsa-miR-466 [57]. However, an increase in hsa-miR-466 transcriptional activity was observed in malignant tumours of the cervix uteri [58].
In our study, increased expression of hsa-miR-466 negatively correlated with the mRNA level of the CKS2 oncogene. CKS2 encodes a regulatory subunit of 2 cyclin-dependent kinases. Increased expression of CKS2 is associated with tumour progression in bladder cancer, hepatocellular carcinoma, breast cancer and GBM [59,60,61,62].
We did not find microRNA-mRNA correlation pairs in the GBM group. However, thirteen expressed miRNAs were specific for GBM. These miRNAs are also very promising for screening panels. It is worth noting that in the TCGA GBM project, there is an extreme lack of miRNA sequencing data. Future studies using next-generation sequencing technology will help uncover new links and mechanisms in GBM oncogenesis.
Previously, studies have been carried out aimed at finding genetic markers for the molecular classification of glial tumours. In the study of Shai R et al. [63], cluster analysis of 170 DEGs made it possible to differentiate the subtypes of astrocytomas, oligodendrogliomas and glioblastomas. However, the number of samples included in the study was relatively small—35 samples of gliomas of various grades of malignancy. In addition, the percentage of errors in predicting survival was relatively high (22%) [63]. Later, a transcriptomic study of 225 samples of glial tumours was carried out. The authors were able to find a relationship between a favourable prognosis and a mutation in IDH1/2, which was most common in G1 tumours. Nonetheless, no subtype-specific genetic signature was found [64]. There are also a number of other research papers devoted to the study of the molecular characteristics of one or two types of glial tumours [65,66,67]. Our study is characterized by a large number of samples and tumour subtypes (939 samples of 5 different subtypes of gliomas were analysed) and an integrative approach, which consists in the analysis of signalling pathways, survival and the search for subtype-specific markers among DEGs and DE miRNAs. The results obtained open up broad prospects in the early diagnosis of certain subtypes of gliomas and in predicting the overall survival of patients using miRNA and mRNA panels. We hope that further experimental studies of expressions subtype-specific miRNAs and mRNAs will confirm the effectiveness of appropriate genetic tests, which will allow clinicians to timely stratify patients depending on the tumour subtype and choose the optimal treatment regimen.
Conclusions
As a result of our study, the molecular genetic characteristics of five glioma subtypes were described: astrocytoma G2, astrocytoma G3, oligodendroglioma G2, oligodendroglioma G3 and GBM G4. We have shown that each subtype has a unique expression profile of genes and miRNAs and has differences in key signalling pathways and regulatory networks of miRNA-RNA interactions. Based on the analysis of differential expression and overall survival, we formed a panel of genes and miRNAs that allows the identification of different glioma subtypes and the evaluation of prognosis in terms of overall survival. The identified regulatory miRNA-mRNA pairs unique to the five glioma subtypes can stimulate the development of new therapeutic approaches based on subtype-specific regulatory mechanisms.
Availability of data and materials
The datasets analysed during the current study are available in the GDC Data Portal repository, https://portal.gdc.cancer.gov.
Abbreviations
- 24-OHC:
-
24S-Hydroxycholesterol
- CRC:
-
Colorectal cancer
- DEG:
-
Differential gene expression
- DE miRNA:
-
Differential miRNA expression
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GBM:
-
Glioblastoma
- GSEA:
-
Gene set enrichment analysis
- miRNA:
-
MicroRNA
- OS:
-
Overall survival
- TCGA:
-
The Cancer Genome Atlas
References
Bauchet L, Ostrom QT (2019) Epidemiology and Molecular Epidemiology. Neurosurg Clin N Am. 30(1):1–16 https://doi.org/10.1016/j.nec.2018.08.010
Arcella A, Limanaqi F, Ferese R et al (2020) Dissecting molecular features of gliomas: genetic loci and validated biomarkers. Int J Mol Sci. 21(2):1–17 https://doi.org/10.3390/ijms21020685
Jiang WD, Ye ZH (2019) Integrated analysis of a competing endogenous RNA network in renal cell carcinoma using bioinformatics tools. Biosci Rep. 39(7):BSR20190996 https://doi.org/10.1042/BSR20190996
Jevšinek Skok D, Hauptman N, Boštjančič E et al (2019) The integrative knowledge base for miRNA-mRNA expression in colorectal cancer. Sci Rep. 9(1):18065 https://doi.org/10.1038/s41598-019-54358-w
Zhang J, Li D, Zhang Y et al (2020) Integrative analysis of mRNA and miRNA expression profiles reveals seven potential diagnostic biomarkers for non-small cell lung cancer. Oncol Rep. 43(1):99–112 https://doi.org/10.3892/or.2019.7407
Wei J, Yin Y, Deng Q et al (2020) Integrative Analysis of MicroRNA and Gene Interactions for Revealing Candidate Signatures in Prostate Cancer. Front Genet. 11:176 https://doi.org/10.3389/fgene.2020.00176
Colaprico A, Silva TC, Olsen C et al (2016) TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44(8):e71 https://doi.org/10.1093/nar/gkv1507
Li R, Qu H, Wang S et al (2018) GDCRNATools: An R/Bioconductor package for integrative analysis of lncRNA, miRNA and mRNA data in GDC. Bioinformatics. 34(14):2515–2517 https://doi.org/10.1093/bioinformatics/bty124
Xu T, Su N, Liu L et al (2018) miRBaseConverter: An R/Bioconductor package for converting and retrieving miRNA name, accession, sequence and family information in different versions of miRBase. BMC Bioinformatics 19(Suppl 19):514 https://doi.org/10.1186/s12859-018-2531-5
Ritchie ME, Phipson B, Wu D et al (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7):e47 https://doi.org/10.1093/nar/gkv007
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4(1):44–57 https://doi.org/10.1038/nprot.2008.211
Szklarczyk D, Gable AL, Lyon D et al (2019) STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47(D1):D607–D613 https://doi.org/10.1093/nar/gky1131
Wang S, Liu X (2019) The UCSCXenaTools R package: a toolkit for accessing genomics data from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq. J Open Source Softw. 4(40):1627 https://doi.org/10.21105/joss.01627
Ru Y, Kechris KJ, Tabakoff B et al (2014) The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res 42(17):e133 https://doi.org/10.1093/nar/gku631
Weng J, Xiao J, Mi Y et al (2018) PCDHGA9 acts as a tumor suppressor to induce tumor cell apoptosis and autophagy and inhibit the EMT process in human gastric cancer. Cell Death Dis 9(2):27 https://doi.org/10.1038/s41419-017-0189-y
Zhang X, Ning Y, Xiao Y et al (2017) MAEL contributes to gastric cancer progression by promoting ILKAP degradation. Oncotarget. 8(69):113331–113344 https://doi.org/10.18632/oncotarget.22970
Matsuura K, Sawai H, Ikeo K et al (2017) Genome-wide association study identifies TLL1 variant associated with development of hepatocellular carcinoma after eradication of hepatitis C virus infection. Gastroenterology. 152(6):1383–1394 https://doi.org/10.1053/j.gastro.2017.01.041
Seifert M, Peitzsch C, Gorodetska I et al (2019) Network-based analysis of prostate cancer cell lines reveals novel marker gene candidates associated with radioresistance and patient relapse. PLoS Comput Biol. 15(11):e1007460 https://doi.org/10.1371/journal.pcbi.1007460
Porsch M, Özdemir E, Wisniewski M et al (2019) Time resolved gene expression analysis during tamoxifen adaption of MCF-7 cells identifies long non-coding RNAs with prognostic impact. RNA Biol. 16(5):661–674 https://doi.org/10.1080/15476286.2019.1581597
Park HB, Kim JW, Baek KH (2020) Regulation of Wnt Signaling through ubiquitination and deubiquitination in cancers. Int J Mol Sci. 21(11):E3904. https://doi.org/10.3390/ijms21113904
Guan C, He L, Chang Z et al (2020) ZNF774 is a potent suppressor of hepatocarcinogenesis through dampening the NOTCH2 signaling. Oncogene. 39(8):1665–1680 https://doi.org/10.1038/s41388-019-1075-0
Delma CR, Thirugnanasambandan S, Srinivasan GP et al (2019) Fucoidan from marine brown algae attenuates pancreatic cancer progression by regulating p53 – NFκB crosstalk. Phytochemistry. 167:112078 https://doi.org/10.1016/j.phytochem.2019.112078
Yang WJ, Wang HB, Da Wang W et al (2020) A network-based predictive gene expression signature for recurrence risks in stage II colorectal cancer. Cancer Med. 9(1):179–193 https://doi.org/10.1002/cam4.2642
Mortazavi D, Sharifi M (2018) Antiproliferative effect of upregulation of hsa-let-7c-5p in human acute erythroleukemia cells. Cytotechnology. 70(6):1509–1518 https://doi.org/10.1007/s10616-018-0241-5
Hou N, Han J, Li J et al (2014) MicroRNA profiling in human colon cancer cells during 5-fluorouracil-induced autophagy. PLoS One. 9(12):e114779 https://doi.org/10.1371/journal.pone.0114779
Wang G, Mao W, Zheng S et al (2009) Epidermal growth factor receptor-regulated miR-125a-5p - A metastatic inhibitor of lung cancer. FEBS J. 276(19):5571–5578 https://doi.org/10.1111/j.1742-4658.2009.07238.x
Scott GK, Goga A, Bhaumik D et al (2007) Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 282(2):1479–1486 https://doi.org/10.1074/jbc.M609383200
Jia HY, Wang YX, Yan WT et al (2012) MicroRNA-125b functions as a tumor suppressor in hepatocellular carcinoma cells. Int J Mol Sci. 13(7):8762–8774 https://doi.org/10.3390/ijms13078762
Morelli E, Leone E, Cantafio MEG et al (2015) Selective targeting of IRF4 by synthetic microRNA-125b-5p mimics induces anti-multiple myeloma activity in vitro and in vivo. Leukemia. 29(11):2173–2183 https://doi.org/10.1038/leu.2015.124
Yuan M, Da Silva AL, Arnold A et al (2018) MicroRNA (miR) 125b regulates cell growth and invasion in pediatric low grade glioma. Sci Rep. 8(1):12506 https://doi.org/10.1038/s41598-018-30942-4
Sun YM, Lin KY, Chen YQ (2013) Diverse functions of miR-125 family in different cell contexts. J Hematol Oncol. 6:6 https://doi.org/10.1186/1756-8722-6-6
Wu N, Lin X, Zhao X et al (2013) MiR-125b acts as an oncogene in glioblastoma cells and inhibits cell apoptosis through p53 and p38MAPK-independent pathways. Br J Cancer. 109(11):2853–2863 https://doi.org/10.1038/bjc.2013.672
Jin Z, Xu S, Yu H et al (2013) MiR-125b inhibits connexin43 and promotes glioma growth. Cell Mol Neurobiol. 33(8):1143–1148 https://doi.org/10.1007/s10571-013-9980-1
Yang J, Liu W, Lu X et al (2015) High expression of small GTPase Rab3D promotes cancer progression and metastasis. Oncotarget. 6(13):11125–11138 https://doi.org/10.18632/oncotarget.3575
Jin T, Liu M, Liu Y et al (2020) Lcn2-derived Circular RNA (hsa_circ_0088732) Inhibits cell apoptosis and promotes EMT in glioma via the miR-661/RAB3D Axis. Front Oncol. 10:170 https://doi.org/10.3389/fonc.2020.00170
Dai W, He J, Zheng L et al (2019) miR-148b-3p, miR-190b, and miR-429 regulate cell progression and act as potential biomarkers for breast cancer. J Breast Cancer. 22(2):219–236 https://doi.org/10.4048/jbc.2019.22.e19
Han JY, Shin ES, Lee YS et al (2013) A genome-wide association study for irinotecan-related severe toxicities in patients with advanced non-small-cell lung cancer. Pharmacogenomics J. 13(5):417–422 https://doi.org/10.1038/tpj.2012.24
Wang YN, Chen ZH, Chen WC (2017) Novel circulating microRNAs expression profile in colon cancer: A pilot study. Eur J Med Res. 22(1):51 https://doi.org/10.1186/s40001-017-0294-5
Chen Y, Huang S, Wu B et al (2017) Transforming growth factor-β1 promotes breast cancer metastasis by downregulating miR-196a-3p expression. Oncotarget. 8(30):49110–49122 https://doi.org/10.18632/oncotarget.16308
Feng Z, Zhang L, Zhou J et al (2017) mir-218-2 promotes glioblastomas growth, invasion and drug resistance by targeting CDC27. Oncotarget. 8(4):6304–6318 https://doi.org/10.18632/oncotarget.13850
He H, Li W, Yan P et al (2018) Identification of a recurrent LMO7-BRAF fusion in papillary thyroid carcinoma. Thyroid. 28(6):748–754 https://doi.org/10.1089/thy.2017.0258
Nakamura H, Hori K, Tanaka-Okamoto M et al (2011) Decreased expression of LMO7 and its clinicopathological significance in human lung adenocarcinoma. Exp Ther Med. 2(6):1053–1057 https://doi.org/10.3892/etm.2011.329
Tanaka-Okamoto M, Hori K, Ishizaki H et al (2009) Increased susceptibility to spontaneous lung cancer in mice lacking LIM-domain only 7. Cancer Sci. 100(4):608–616 https://doi.org/10.1111/j.1349-7006.2009.01091.x
Kvarnbrink S, Karlsson T, Edlund K et al (2015) LRIG1 is a prognostic biomarker in non-small cell lung cancer. Acta Oncol (Madr). 54(8):1113–1119 https://doi.org/10.3109/0284186X.2015.1021427
Mullany LE, Herrick JS, Sakoda LC et al (2018) miRNA involvement in cell cycle regulation in colorectal cancer cases. Genes and Cancer. 9(1-2):860–872 https://doi.org/10.18632/genesandcancer.167
Ulivi P, Canale M, Passardi A et al (2018) Circulating plasma levels of miR-20b, miR-29b and mir-155 as predictors of bevacizumab efficacy in patients with metastatic colorectal cancer. Int. J. Mol. Sci. 19(1):307 https://doi.org/10.3390/ijms19010307
Espiard S, Knape MJ, Bathon K et al (2018) Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight. 3(8):e98296 https://doi.org/10.1172/jci.insight.98296
Wang Y, Wang B, Zhou H et al (2020) MicroRNA-384 inhibits the progression of papillary thyroid cancer by targeting PRKACB. Biomed Res Int. 2020:4983420 https://doi.org/10.1155/2020/4983420
Chen Y, Gao Y, Tian Y et al (2013) PRKACB is Downregulated in non-small cell lung cancer and exogenous PRKACB inhibits proliferation and invasion of LTEP-A2 cells. Oncol Lett. 5(6):1803–1808 https://doi.org/10.3892/ol.2013.1294
Niyazi M, Pitea A, Mittelbronn M et al (2016) A 4-miRNA signature predicts the therapeutic outcome of glioblastoma. Oncotarget. 7(29):45764–45775 https://doi.org/10.18632/oncotarget.9945
Uhr K, Prager-Van der Smissen WJC, Heine AAJ et al (2019) MicroRNAs as possible indicators of drug sensitivity in breast cancer cell lines. PLoS One 14(5):e0216400 https://doi.org/10.1371/JOURNAL.PONE.0216400
Zhou X, Zhang Z, Liang X (2020) Regulatory network analysis to reveal important miRNAs and genes in non-small cell lung cancer. Cell J. 21(4):459–466 https://doi.org/10.22074/cellj.2020.6281
Silvente-Poirot S, Dalenc F, Poirot M (2018) The effects of cholesterol-derived oncometabolites on nuclear receptor function in cancer. Cancer Res. 78(17):4803–4808 https://doi.org/10.1158/0008-5472.CAN-18-1487
Han M, Wang S, Yang N et al (2020) Therapeutic implications of altered cholesterol homeostasis mediated by loss of CYP46A1 in human glioblastoma. EMBO Mol Med. 12(1):e10924 https://doi.org/10.15252/emmm.201910924
Sun Y, Zhou J, Shi L et al (2019) MicroRNA-466 inhibits cell proliferation and invasion in osteosarcoma by directly targeting insulin receptor substrate 1. Mol Med Rep. 19(4):3345–3352 https://doi.org/10.3892/mmr.2019.9956
Jia C, Tang D, Sun C et al (2018) MicroRNA-466 inhibits the aggressive behaviors of hepatocellular carcinoma by directly targeting metadherin. Oncol Rep. 40(6):3890–3898 https://doi.org/10.3892/or.2018.6763
Li G, Zhang Y, Mao J et al (2019) LncRNA TUC338 is overexpressed in prostate carcinoma and downregulates miR-466. Gene. 707:224–230 https://doi.org/10.1016/j.gene.2019.05.026
Zhou LL, Shen Y, Gong JM et al (2017) MicroRNA-466 with tumor markers for cervical cancer screening. Oncotarget. 8(41):70821–70827 https://doi.org/10.18632/oncotarget.19992
Chen R, Feng C, Xu Y (2011) Cyclin-dependent kinase-associated protein Cks2 is associated with bladder cancer progression. J Int Med Res. 39(2):533–540 https://doi.org/10.1177/147323001103900222
Lin L, Fang Z, Lin H et al (2016) Depletion of Cks1 and Cks2 expression compromises cell proliferation and enhance chemotherapy-induced apoptosis in HepG2 cells. Oncol Rep. 35(1):26–32 https://doi.org/10.3892/or.2015.4372
Wang J, Xu L, Liu Y et al (2014) Expression of cyclin kinase subunit 2 in human breast cancer and its prognostic significance. Int J Clin Exp Pathol. 7(12):8593–8601
Huang N, Wu Z, Hong H et al (2019) Overexpression of CKS2 is associated with a poor prognosis and promotes cell proliferation and invasion in breast cancer. Mol Med Rep. 19(6):4761–4769 https://doi.org/10.3892/mmr.2019.10134
Shai R, Shi T, Kremen TJ et al (2003) Gene expression profiling identifies molecular subtypes of gliomas. Oncogene. 22(31):4918–4923 https://10.1038/sj.onc.1206753
Yan W, Zhang W, You G et al (2012) Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol. 14(12):1432–1440 https://10.1093/neuonc/nos263
Liu Z, Yao Z, Li C et al (2011) Gene expression profiling in human high-grade astrocytomas. Comp Funct Genomics. 2011:245137 https://doi.org/10.1155/2011/245137
Bo L, Wei B, Li C et al (2017) Identification of potential key genes associated with glioblastoma based on the gene expression profile. Oncol Lett. 14(2):2045–2052 https://doi.org/10.3892/ol.2017.6460
Xu Y, Geng R, Yuan F, Sun Q, Liu B, Chen Q (2019) Identification of differentially expressed key genes between glioblastoma and low-grade glioma by bioinformatics analysis. PeerJ. 7:e6560 https://doi.org/10.7717/peerj.6560
Acknowledgements
Not applicable.
Funding
We received no financial support from grants of pharmaceutical or other commercial organizations.
Author information
Authors and Affiliations
Contributions
DYG was responsible for methodology, software, formal analysis and investigation. AAP was responsible for writing original manuscript and data visualization. NNT was responsible for supervision, review and editing manuscript. EER was responsible for conceptualization and writing original manuscript. AMN was responsible for data visualization. OIK was responsible for conceptualization and project administration. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors report no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Online Resource 1.
Volcano plot of differential gene expression in anaplastic astrocytoma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC < -1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” genes with reduced/increased expression.
Additional file 2: Online Resource 2.
Volcano plot of differential gene expression in diffuse oligodendroglioma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC <-1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” genes with reduced/increased expression.
Additional file 3: Online Resource 3.
Volcano plot of differential gene expression in anaplastic oligodendroglioma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC < -1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” genes with reduced/increased expression.
Additional file 4: Online Resource 4.
Volcano plot of differential gene expression in glioblastoma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC < -1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” genes with reduced/increased expression.
Additional file 5: Online Resource 5.
Genetic ontology of DEGs specific to anaplastic astrocytoma. (A) Biological process; (B) Cellular component; (C) Molecular function; (D) Pathways.
Additional file 6: Online Resource 6.
Genetic ontology of DEGs specific to diffuse oligodendroglioma. (A) Biological process; (B) Cellular component; (C) Molecular function; (D) Pathways.
Additional file 7: Online Resource 7.
Genetic ontology of DEGs specific to anaplastic oligodendroglioma. (A) Biological process; (B) Cellular component; (C) Molecular function; (D) Pathways.
Additional file 8: Online Resource 8.
Genetic ontology of DEGs specific to glioblastoma. (A) Biological process; (B) Cellular component; (C) Molecular function; (D) Pathways.
Additional file 9: Online Resource 9.
Volcano plot of differential miRNA expression in anaplastic astrocytoma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC <-1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” miRNAs with reduced/increased expression.
Additional file 10: Online Resource 10.
Volcano plot of differential miRNA expression in diffuse oligodendroglioma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC < -1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” miRNAs with reduced/increased expression.
Additional file 11: Online Resource 11.
Volcano plot of differential miRNA expression in anaplastic oligodendroglioma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC < -1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” miRNAs with reduced/increased expression.
Additional file 12: Online Resource 12.
Volcano plot of differential miRNA expression in glioblastoma. The abscissa axis represents the logFC value, and the ordinate axis represents –log10 (P-value with FDR correction). Genes with logFC > 1 and logFC <-1 (with a p-value <0.05) are marked in red and green, respectively. The labels indicate the “top 10” miRNAs with reduced/increased expression.
Additional file 13: Online Resource 13.
Interactome analysis of genes associated with overall survival in anaplastic astrocytoma clustered according to the p-value of protein-protein interactions.
Additional file 14: Online Resource 14.
Interactome analysis of genes associated with overall survival in diffuse oligodendroglioma clustered according to the p-value of protein-protein interactions.
Additional file 15: Online Resource 15.
Interactome analysis of genes associated with overall survival in anaplastic oligodendroglioma clustered according to the p-value of protein-protein interactions.
Additional file 16: Online Resource 16.
Interactome analysis of genes associated with overall survival in glioblastoma, clustered according to p-value of protein-protein interactions.
Additional file 17: Online Resource 17.
Scheme of interactions of miRNAs and the mRNA of genes associated with survival in the studied samples of anaplastic astrocytoma. The red colour denotes DE miRNAs, and the green colour denotes DEGs.
Additional file 18: Online Resource 18.
Scheme of interactions of miRNAs and the mRNA of genes associated with survival in the studied samples of diffuse oligodendroglioma. The red colour denotes DE miRNAs, and the green colour denotes DEGs.
Additional file 19: Online Resource 19.
Scheme of interactions of miRNAs and the mRNA of genes associated with survival in the studied samples of anaplastic oligodendroglioma. The red colour denotes DE miRNAs, and the green colour denotes DEGs.
Additional file 20: Online Resource 20.
Subtype-specific DEGs associated with overall survival.
Additional file 21: Online Resource 21.
Subtype-specific DE miRNAs associated with overall survival.
Additional file 22: Online Resource 22.
Analysis of differential gene expression in diffuse astrocytoma.
Additional file 23: Online Resource 23.
Analysis of differential gene expression in anaplastic astrocytoma.
Additional file 24: Online Resource 24.
Analysis of differential gene expression in diffuse oligodendroglioma.
Additional file 25: Online Resource 25.
Analysis of differential gene expression in anaplastic oligodendroglioma.
Additional file 26: Online Resource 26.
Analysis of differential gene expression in glioblastoma.
Additional file 27: Online Resource 27.
Analysis of differential miRNAs expression in diffuse astrocytoma.
Additional file 28: Online Resource 28.
Analysis of differential miRNAs expression in anaplastic astrocytoma.
Additional file 29: Online Resource 29.
Analysis of differential miRNAs expression in diffuse oligodendroglioma.
Additional file 30: Online Resource 30.
Analysis of differential miRNAs expression in anaplastic oligodendroglioma.
Additional file 31: Online Resource 31.
Analysis of differential miRNAs expression in glioblastoma.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gvaldin, D.Y., Pushkin, A.A., Timoshkina, N.N. et al. Integrative analysis of mRNA and miRNA sequencing data for gliomas of various grades. Egypt J Med Hum Genet 21, 73 (2020). https://doi.org/10.1186/s43042-020-00119-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-020-00119-8