
Abstract
Background
Melanomas are highly aggressive in nature known for metastasis and death. Melanocytes that gave rise to melanomas are neural crest progenitor cells. Our research was primarily concerned with uveal melanoma (UM) and cutaneous melanoma (CM), respectively. Although they both have the same melanocytic origin, the biology of their respective is different.
Aim
The aim of our study was to recognize the common differentially expressed genes (DEGs) between UM and CM.
Methodology
The gene expression profile was downloaded from the GEO and analyzed by GEO2R to recognize DEGs. By applying DAVID, GO, and KEGG, pathway enrichment analysis was performed. PPI of these DEGs was analyzed using STRING and visualized by Cytoscape and MCODE. Further, we utilized HPA and GEPIA to obtain Kaplan–Meier graph for survival analysis in order to assess the prognostic value of hub genes.
Results
We examined the UM and CM datasets and discovered three common upregulated and eight downregulated DEGs based on computational analysis. HMGCS1 and ELOVL5 were shown to be enriched in a variety of altered molecular pathways and pathways in cancer. Overexpression of HMGCS1 and ELOVL5 was linked to a poor prognosis in CM.
Conclusion
Computational evaluation found that HMGCS1 and ELOVL5 were upregulated in both melanomas. Enrichment analysis showed that these genes are involved in cancer metabolism pathway and associated with poor prognosis in CM. However, the molecular study of these genes in UM is limited. Therefore, a better understanding of the cancer metabolism pathways should be carried to pave the way for clinical benefits.
Similar content being viewed by others


Introduction
Melanoma is an aggressive tumor arising from uncontrolled proliferation of pigment-producing cells called melanocytes [1]. These cells originate from neural crest progenitors during embryonic development, giving rise to cutaneous and choroidal cells with distinct characteristics [2]. Cutaneous melanoma (CM) develops from melanocytes in the deep epidermal layer, while uveal melanoma (UM) involves melanocytes in the uvea (iris, ciliary body, and choroid). Shared pathways in melanocyte proliferation are implicated in both types. Various risk factors contribute to melanoma, including age, sex, genetics, phenotypic predisposition, work environment, and dermatological diseases [3]. Ultraviolet radiation is a major risk factor for cutaneous melanoma, linked to strong and intermittent sunlight exposure leading to DNA alterations [4]. However, data on UV's role in uveal melanoma are limited and conflicting.UM and CM, despite a common origin, differ in metastatic biology. UM primarily spreads hematogenously, with the liver being the predominant metastatic site in over 90% of cases. In contrast, CM can metastasize through hematogenic and lymphatic pathways, affecting organs like the lung, lymph nodes, brain, and skin [5, 6]. CM's aggressive metastatic nature distinguishes it as more formidable than UM of comparable histogenesis.
The etiology of both uveal and cutaneous melanoma is complex and heterogeneous. In CM, B. Uzdensky et al., 2013, stated that mutations were observed in the Ras/Raf/MEK pathway, which is a key regulator of cell proliferation and survival [7]. Signature mutations in Ras or Raf proteins promoted cell proliferation, tumor invasion, and metastasis indefinitely. Permanent cell proliferation, apoptosis resistance, and malignant transformation are all caused by mutations in the NRAS and BRAF genes. The activating BRAF mutations are found to be important in the pathophysiology of melanoma. It has been demonstrated that mutated BRAf irreversibly activates ERK and increases proliferation [7]. Additionally, various studies have also revealed that cutaneous melanoma patients frequently harbor alterations in NF1, TP53, and CDKN2A, where CDKN2A mutations were susceptible in both CM and UM [8]. Katopodis et al. specified one of the key molecular pathways observed in uveal melanoma is PI3K/Akt/PTEN which is involved in cell proliferation [9]. BAP1 is one of the most significant mutations recognized in uveal melanoma [10]. And mutations in GNAQ and GNA11 are the most frequent mutations observed in UM. At the cellular level, cutaneous and uveal melanomas share the certain molecular profile as several studies have indicated that in both melanoma types, the mitogen-activated protein kinase (MAPK) pathway is involved, and it regulates melanogenesis. In addition, the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathways that regulate cell growth, differentiation, migration, and survival, and angiogenesis and metabolism are also known to undergo dysregulation leading to genetic alterations and poor treatment outcomes in both CM and UM. Also, the microphthalmia (MITF) transcription factor which is critical to the growth development and progression of melanocytes and melanoma [11] is known to be biochemically targeted by the c-kit signaling pathway implying a link to proliferation [12]. Ortega et al. have revealed that mutations in the telomerase reverse transcriptase (TERT) gene and EIF1AX have been found to be common with both cutaneous melanoma and ocular melanoma [3].
There has been a revolution in understanding the molecular complexity of melanoma biology and tremendous advancements in treatment over the previous decade for CM but the same for UM has been gradual. In this present study, the utilization of computational networking analysis along with various online bioinformatic tools has given us with means to investigate more about the relationship between uveal melanoma and cutaneous melanoma. The aim was to recognize the common differentially expressed genes (DEGs) dysregulated genes, the cancer pathways, and the metabolic pathways which may be common to both these melanoma tumor types.
Material and methods
Data procurement
The gene expression profile datasets used in this investigation were collected from NCBI's database-Gene Expression Omnibus (GEO) [https://www.ncbi.nlm.nih.gov/geo/]. The database yielded a total of 43,707 series related to human cutaneous melanoma and 1227 series related to human uveal melanoma. The gene expression profiles GSE15605 [13] and GSE44295 [14] were selected after careful contemplation for cutaneous and uveal melanoma, respectively. Among the two, GSE15605 was based on platform GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array while GSE44295 was based on the platform GPL6883 Illumina HumanRef-8 v3.0 expression bead chip. All of the data are publicly available online, and no trials on humans or animals were done by any of the authors for this study. As public records were evaluated, no ethical approval was required for this study.
Data processing of DEGs
An online freely available tool, GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r/), was used to identify the DEGs for uveal and cutaneous melanoma by comparing the tumor with normal specimens. DEGs were defined as genes that satisfied the cutoff criterion of adjusted P0.05 and | logFC|> 1.0. For each dataset, statistical analysis was done, and the Venn diagram online tool (bioinformatics.psb.ugent.be/webtools/Venn) was employed to find the common DEGs shared by both uveal and cutaneous melanomas.
PPI network construction and module evaluation
Establishment of PPI Networks and Module Analysis. Using the STRING web-based tool (v11.0, 3) (https://string-db.org/) to analyze the association among the DEGs from the obtained datasets, we built a PPI (protein–protein interaction) network. The network was visualized using Cytoscape software [15] and major gene clusters were identified. Cluster two, with maximum number of genes, was selected for further analysis, and Centiscape plug-in on Cytoscape was used to identify the hub genes. Genes with maximum MCC score were regarded as the hub genes and studied.
Gene ontology analysis
Gene Ontology is a frequently used approach for linking genes, gene products, and sequences to their biological processes. Gene functions are divided into three categories: biological process (BP), molecular function (MF), and cellular component (CC). KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database which can be used to understand genomic sequences and other high-throughput data in a biological context. Both these analyses were done using the ShinyGO database [16] which is a bioinformatics data repository with genomes and pathways information for over 315 plant and animal species, based on Ensembl, Ensembl plants, Ensembl Metazoa, STRING, and KEGG databases. P value < 0.05 was considered significant.
Survival analysis of key genes
The Human Protein Atlas [17] is an open access software with information in protein upon tissue, brain section, single cell type, tissue cell type, metabolism, etc. We used HPA database to obtain the Kaplan–Meier plots of the key genes, wherein p value < 0.05 was considered significant. The Km plots were used to analyze their role as prognostic markers for cancer.
Immunohistochemical evaluation
The Human Protein Atlas (HPA, https://www.proteinatlas.org/) database was used to obtain immunohistochemical staining to verify the protein expression level of key genes in cutaneous melanoma. However, data for uveal melanoma were not available in any of the free databases.
Results
Identification of common genes
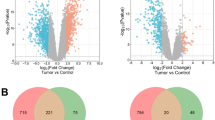
GSE15605 and GSE44295 (Supplementary Table 1) are two datasets retrieved from the GEO repository that were then subjected to data analysis in accordance with our inclusion criteria. The P < 0.05 and |logFC|> 1 criterion was used to retrieve DEGs from the GEO database. The overlap between the two datasets is seen in the Venn diagram (Fig. 1). Four hundred and ninety-two DEGs were significantly similar in expression between the two groups and were included in the study.

Venn diagram showcasing 492 common differentially expressed genes between uveal and cutaneous melanomas
PPI network construction and module evaluation
Employing STRING online tool, the DEG PPI network was constructed (Supplementary Fig. 1). Cluster 2 was identified using MCODE in Cytoscape (Fig. 2A), and it displayed significant level of common DEGs (Supplementary Table 2). It was considered as the most important module and was selected. Further 3 upregulated and 8 downregulated genes (Fig. 2B) were identified based on their log Fc values. Following calculations by Centiscape, screening of hub genes using maximum MCC score was done (Supplementary Table 3).

A STRING PPI network as obtained for the genes present in the cluster 2. B Cluster 2 genes, where blue color denotes downregulated genes and red color denotes the upregulated genes in both uveal and cutaneous melanomas
Functional and pathway enrichment
Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used to perform functional and pathway enrichment analysis on cluster 2 DEGs to better understand their biological categorization. According to the results of the GO analysis, DEGs were primarily involved in metabolic pathways such as fatty acid metabolism, lipid biosynthetic and metabolic process, semaphoring-plexin signaling pathway (Fig. 3A).

A Gene Ontology for the cluster 2 genes as obtained using ShinyGO (FDR = 0.01). B Top 10 KEGG Pathways obtained for the cluster 2 genes
Furthermore, the findings of the KEGG pathway study revealed that DEGs were shown to be prominent in the cancer metabolism pathway, fatty acid biosynthesis, and ErbB signaling pathway, Th 17 cell differentiation among other molecular pathways (Fig. 3B).
Survival analysis
The Kaplan–Meier plot was used for survival analysis to evaluate the prognostics significance of hub genes in cutaneous melanoma. High expression of HMGCS1 and ELOVL5 was shown to be related with a poor prognosis in cutaneous melanoma. (Fig. 4A, B).

Km Plots for A HMGCS1 and B ELOVL-5 expression in melanoma obtained from the Human Protein Atlas showcasing regressed survival rates in patients with high expression levels of these genes
Immunohistochemical expression
ELOVL5 AND HMGCS1 immunohistochemical staining was shown to be positive in cutaneous melanoma tissues. It shows that these genes were considerably expressed in cutaneous melanoma tumor tissues (Fig. 5A, B). Although, as previously stated, data for uveal melanoma were not available, this could be owing to a limited number of patient samples.

A Strong cytoplasmic and membranous immunohistochemical expression of HMGCS1 in case of cutaneous melanoma. B Cytoplasmic over expression of ELOVL5 in a representative case of cutaneous melanoma
Discussion
Melanoma is a malignancy of melanocytes and is known for its remarkable ability to metastasize rapidly. Cutaneous melanoma (CM) and uveal melanoma (UM) are two common forms of melanoma that arise from the same embryonic origin but the etiopathogenesis and biological characteristics of these melanomas, however, are highly distinct. Even though they both have same melanocytic origins, the biology of their respective metastasis is different. The goal of our research was to find differentially expressed genes (DEGs) in UM and CM.
Two commonly upregulated hub genes are HMGCS1 AND ELOVL5. Several studies have indicated that HMGCS1 and ELOVL5 to be oncogenic in nature and are found to be involved in different cancer and metabolic pathways [18,19,20,21,22,23].
3-Hydroxy 3- methylglutaryl- CoA synthase 1 (HMGCS1) is known to be potential regulatory node of the mevalonate pathway which is further involved in tumorigenesis by positively regulating the cell proliferation, migration, invasion, and metastasis of tumor cells [18]. HMGCS1 overexpression has been observed in various cancers like breast cancer, gastric cancer, renal cancer, colon cancer, and prostate cancer. Shengzhou et al. have demonstrated that the suppression of HMGCS1 could completely reduce the EGF-induced proliferation of colon cancer cells indicating HMGCS1 as a novel biomarker.
Elongation of very long chain fatty acid-like (ELOVL5) is necessary to produce C20-22 PUFAs. It plays significant function in the synthesis of fatty acid elongation [20] influenced cell migration, cell–cell interactions, and MMP’s synthesis [21]. Substantial evidence suggests that ELOVL5 expression is commonly deregulated in cancers. It has been found to be overexpressed in hepatocellular carcinoma [22] lung cancer [23], and breast cancer; nevertheless [23], many experimental models have demonstrated ELOVL5 to be extremely low in certain cancers; therefore [21], further research on the understanding of this gene in cancer metastasis is warranted.
The upregulation of these genes in several carcinoma suggests that ELOVL5 and HMGCS1 might play an important role in the pathogenesis of CM suggesting, indicating a key role for these novel genes, suggesting that they can be considered as potential therapeutic targets.
Our study reveals a network-based strategy to screen genes and understand multiple metabolic pathways responsible for both CM and UM using data mining and bioinformatic analysis approaches. Although due to the limited number of patients and difficulty of access to tissue, progress in researching and identifying genes for uveal melanoma is limited. The study of cell differentiation pathways and genes, as well as numerous biochemical processes, can assist in the early diagnosis and treatment of melanoma [24]. Therefore, hub genes implicated in the advancement of CM and UM may be investigated further, which might aid in the development of prognostic and therapeutic techniques to cure UM using concepts like those used in CM.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Change history
24 July 2024
A Correction to this paper has been published: https://doi.org/10.1186/s43042-024-00554-x
References
Cichorek M, Wachulska M, Stasiewicz A, Tymińska A (2013) Skin melanocytes: biology and development. Postepy dermatologii i alergologii 30(1):30–41. https://doi.org/10.5114/pdia.2013.33376
Hoefsmit EP, Rozeman EA, Van TM, Dimitriadis P, Krijgsman O, Conway JW, Da Silva IP, Van Der Wal JE, Ketelaars SLC, Bresser K, Broeks A, Kerkhoven R, Reeves J, Warren S, Kvistborg P, Scolyer RA, Kapiteijn E, Peeper DS, Long GV, Blank CU (2020) Comprehensive analysis of cutaneous and uveal melanoma liver metastases. J ImmunoTher Cancer. https://doi.org/10.1136/jitc-2020-001501
Ortega MA, Fraile-Martínez S, García-Honduvilla N, Coca S, Álvarez-Mon M, Buján J, Teus MA (2020) Update on uveal melanoma: translational research from biology to clinical practice (review). Int J Oncol. https://doi.org/10.3892/ijo.2020.5140
Wäster P, Eriksson I, Vainikka L, Rosdahl I, Öllinger K (2016) Extracellular vesicles are transferred from melanocytes to keratinocytes after UVA irradiation. Sci Rep. https://doi.org/10.1038/srep27890
Ibrahim N, Haluska FG (2009) Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol 4:551–579. https://doi.org/10.1146/annurev.pathol.3.121806.151541
Materin MA (2011) The relationship between cutaneous and uveal melanoma. Retina Today
Uzdensky AB (2013) Signal transduction in human cutaneous melanoma and target drugs. https://doi.org/10.2174/1568009611313080004
Kannengiesser C, Avril MF, Spatz A, Laud K, Lenoir GM, Bressac-de-Paillerets B (2003) CDKN2A as a uveal and cutaneous melanoma susceptibility gene. Genes Chromosom Cancer 38(3):265–268. https://doi.org/10.1002/gcc.10286
Katopodis P, Khalifa MS, Anikin V (2021) Molecular characteristics of uveal melanoma and intraocular tumors. Oncol Lett 21(1):9. https://doi.org/10.3892/ol.2020.12270
van der Kooij MK, Speetjens FM, van der Burg SH, Kapiteijn E (2019) Uveal versus cutaneous melanoma; same origin. Very Distinct Tumor Types Cancers 11(6):845. https://doi.org/10.3390/cancers11060845
Kawakami A, Fisher DE (2017) The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Investig J Tech Methods Pathol 97(6):649–656. https://doi.org/10.1038/labinvest.2017.9
Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE (1998) MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 391(6664):298–301. https://doi.org/10.1038/34681
Raskin L, Fullen DR, Giordano TJ, Thomas DG, Frohm ML, Cha KB, Ahn J, Mukherjee B, Johnson TM, Gruber SB (2013) Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J Invest Dermatol 133(11):2585–2592. https://doi.org/10.1038/jid.2013.197
Yogen S (2013) Genome-wide gene expression analysis of primary uveal melanoma cells, normal uveal melanocytes, neural crest stem cells, embryonic stem cells and uveal melanoma cell lines MUM2B and OCM1A
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504. https://doi.org/10.1101/gr.1239303
Ge SX, Jung D, Yao R (2020) ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36(8):2628–2629. https://doi.org/10.1093/bioinformatics/btz931
Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lundberg E (2017) A subcellular map of the human proteome. Science 356(6340):3321. https://doi.org/10.1126/science.aal3321
Walsh CA, Akrap N, Garre E, Magnusson Y, Harrison H, Andersson D, Jonasson E, Rafnsdottir S, Choudhry H, Buffa F, Ragoussis J, Ståhlberg A, Harris A, Landberg G (2020) The mevalonate precursor enzyme HMGCS1 is a novel marker and key mediator of cancer stem cell enrichment in luminal and basal models of breast cancer. PLoS ONE 15(7):e0236187. https://doi.org/10.1371/journal.pone.0236187
Zhou S, Xu H, Tang Q, Xia H, Bi F (2020) Dipyridamole enhances the cytotoxicities of trametinib against colon cancer cells through combined targeting of HMGCS1 and MEK pathway. Mol Cancer Ther 19(1):135–146. https://doi.org/10.1158/1535-7163.MCT-19-0413
Zhang JY, Kothapalli KS, Brenna JT (2016) Desaturase and elongase-limiting endogenous long-chain polyunsaturated fatty acid biosynthesis. Curr Opin Clin Nutr Metab Care 19(2):103–110. https://doi.org/10.1097/MCO.0000000000000254
Tripathy S, Jump DB (2013) Elovl5 regulates the mTORC2-Akt-FOXO1 pathway by controlling hepatic cis-vaccenic acid synthesis in diet-induced obese mice. J Lipid Res 54(1):71–84. https://doi.org/10.1194/jlr.M028787
Burns TA, Kadegowda AK, Duckett SK, Pratt SL, Jenkins TC (2012) Palmitoleic (16:1 cis-9) and cis-vaccenic (18:1 cis-11) acid alter lipogenesis in bovine adipocyte cultures. Lipids 47(12):1143–1153. https://doi.org/10.1007/s11745-012-3723-9
Marien E, Meister M, Muley T, Gomez Del Pulgar T, Derua R, Spraggins JM, Van de Plas R, Vanderhoydonc F, Machiels J, Binda MM, Dehairs J, Willette-Brown J, Hu Y, Dienemann H, Thomas M, Schnabel PA, Caprioli RM, Lacal JC, Waelkens E, Swinnen JV (2016) Phospholipid profiling identifies acyl chain elongation as a ubiquitous trait and potential target for the treatment of lung squamous cell carcinoma. Oncotarget 7(11):12582–12597. https://doi.org/10.1863/oncotarget.7179
Paula BheringNogueira A, Drummond-Lage AP, Drummond Pinho Ribeiro G et al (2022) Factors related to delayed diagnosis of cutaneous melanoma in the brazilian public health system. J Canc Educ 37:1253–1259. https://doi.org/10.1007/s13187-022-02176-6
Acknowledgements
Principal Prof. Dinesh Khattar, Kirori Mal College, University of Delhi and Principal Prof. C. Sheela Reddy, Sri Venkateswara College, University of Delhi and SRIVIPRA.
Funding
No funding received.
Author information
Authors and Affiliations
Contributions
P.J and A.P conceived and planned the experiments. T.B, K.K, S. T and K.G and performed network analysis and analyzed the data. T.B, S.T, K.G wrote the manuscript with input from all authors. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: authors Khushneet Kaur and Shreya Taluja were incorrectly affiliated.
Supplementary Information
Additional file 1: Supplementary Figure 1.
Protein-protein Interactions for the 492 targets as obtained from the STRING Network. Supplementary Table 1. GEO datasets chosen for the study obtained from NCBI GEO2R database. Supplementary Table 2. Upregulation and downregulation of cluster 2 genes in uveal and cutaneous melanomas. Supplementary Table 3. Hub and Bottleneck genes identified using degree and betweenness filter respectively.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jayaraj, P., Bhimwal, T., Kaur, K. et al. A bioinformatics approach to reveal common genes and molecular pathways shared by cutaneous melanoma and uveal melanoma. Egypt J Med Hum Genet 25, 56 (2024). https://doi.org/10.1186/s43042-024-00526-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00526-1