
Abstract
Background
Alzheimer disease (AD) is an ailment that disturbs mainly people of old age. The fundamental remedial way to deal with AD depends on the utilization of AChEI. The design of new intense and particular AChEI is critical in drug discovery. In silico technique will be used to solve the above problem. A new method was established to discover novel agents with better biological activity against Alzheimer disease.
Results
A validated model was established in this research to predict the biological activities of some anti-Alzheimer compounds and to design new hypothetical drugs influenced with molecular properties in the derived model; ATS4i, MATS2e, SpMax7_BhS, Energy(HOMO) and Molecular Weight and showed good correlation R2 = 0.936, R2adj = 0.907, Q2cv = 0.88, LOF = 0.0154 and R2ext = 0.881. All the descriptors in the model were in good agreement with the 15 test set predicted values. Five compounds were designed using D35rm as a template with improved activity. The compounds have higher and better binding scores (− 10.1, − 9.4, − 9.3, − 9.1 and − 8.1 all in kcal/mol) than the approved drugs (Donepezil = − 7.4 kcal/mol).
Conclusion
As the outcome, every one of the selected and the designed compounds is created and improved as potential anti-Alzheimer agents. Despite this, the further test examines and in vivo investigations are recommended to assess the method of the activities and other pharmacological impacts on these compounds.
Similar content being viewed by others
Background
Among numerous infections influencing present-day humanities, dementia is quite possibly the most severe health problem. The most well-known type of dementia is AD which is liable for a large percentage of the circumstances. In line with these realities, 40 years to come, a hundred million AD patients may spring up [1]. A decrease in memory is one out of the numerous characteristics of AD which result in neuronal atrophy [2]. The existing pharmacological administration of communication and emotional signs of dementia (BPSD) offers restricted viability, related to genuine adverse effects bringing about an enhanced danger of death [3].
In the cure of AD, quite a lot of scientific trials have made known that ACHEIs are favourable treatments [4, 5]. Tacrine, galantamine, rivastigmine and donepezil are some ACHEIs presently used, but their clinical usefulness is inadequate since the inadequate number of available drugs, their low effectiveness and numerous unwanted side effects such as hepatotoxicity [6]; therefore, AD remains incurable [7,8,9], to increase their activity and lessen adverse side effects [10], and there is need to synthesis and study new compounds as ACHEIs is required [11].
Computational techniques are on a fundamental level similar to high-throughput separating both target and ligand structure, and data are required. The extending highlights of 3D ligand-based pharmacophore screening and pharmacokinetic inspect, improve the lead atoms choice with ligand viability [12, 13].
The present study aimed to identify and validate that N-aryl derivatives bind to the energetic site of ChEIs [14]. Theoretical methods have been effectively used to recognize the necessary structural structures for their selective inhibitory activity. The quantitative structure–activity relationships (QSAR) approaches have the capability of diminishing considerably the time and exertion needed for the disclosure of the different treatments [15]. Molecular descriptors that address the variety of the underlying properties of the particles are significant progress in building the QSAR models [15]. Subsequently, the utilization of QSAR in the improvement of a hypothetical model to calculate the biological activities of a set of compounds is a vital strategy used in the QSAR methodology and is found in the literature [16].
Also, the following approaches were used to determine the efficacy of the ligand inhibitor molecules: design of molecules, 3D pharmacophore, molecular docking, and ADMET studies are essential approaches to determining ligand inhibitors.
Methods
Data set, generation of the molecular structures and descriptor calculation
The QSAR studies contain 75 N-aryl derivatives with the anti-Alzheimer activity that were selected from the literature [17,18,19,20]. For this study, ChemDraw Ultra version 12.0, Spartan’14 version 1.1.4 and PaDEL-Descriptor Software, version 2.18 were used, anti-Alzheimer activity values of the designated molecules were described in IC50 (µmol); it was transformed to the inverse of IC50 and then to negative Log of IC50 [21]. 3D of the structures was obtained from the drawn 2D molecular structures by ChemDraw software; optimization and DFT calculations were made on the molecules by the Spartan’14 software and B3LYP/6-31G** basis set which yielded several molecular properties [22].
Data pre-treatment, splitting and descriptors change
All descriptors segments with perpetual and with a difference not exactly 0.001 were removed from the descriptor pool. Correspondence scrutiny was done on any descriptor of any pair with a relationship more prominent than 0.8, which was disposed of. The Modified K-mediod1.2 [23] was utilized to share the data. Descriptors in the model with great positive or negative assessment information of the training set were changed by standardization [24] utilizing the equation under
where Pn is the standardized molecular properties, Pmax is the maximum value of the property section, and Pmin is the least value in the column.
Model molecular properties collection
An algorithm in Material Studio, version 7.0 best describes the difference in activity of the studied molecules that were employed to choose a grouping of descriptors [25]. The algorithm has some merits; one of them is the production of several models at a time [26].
Model development, selection and QSAR model validation
The Molegro Data Modeler programming was used to build the model and export into the Molegro worksheet, the descriptors and activities of the compounds. The selection of models built was done dependent on the conditions of R2, Q2, and R2pred [27, 28].
Mean effect and variance inflation factor (VIF)
The mean effect was restrained towards activities of the produced model and the impact of the calculated descriptors.
where βp modifies the molecular properties p's coefficient, Dp modifies each estimation of framework property in the preparation set, and q adjusts with the count of model properties present and r represents the count of particles utilized as preparing set [27, 28]. Variance inflation factor (VIF) of multi-co-linearity between molecular properties blends selected by the algorithm was assessed. In Eq. (3), R_ip^2 is the relationship coefficient of the different relapse between the property I and the excess j molecular properties in the model [28].
Models applicability domain (ADS′)
The estimation with a given consistency which a model make defines the AD′ of the response and chemical space [29, 30]. Those compounds that fall within the AD′ can be considered dependable if only the model can be positioned to use for selecting compounds, which means AD′ is defined and predicted [31]. Arrangement of a hat matrix H maps the vector of experimental values to the vector of fitted values defined by the leverage method which was used for AD′ in this work to build models [32]. The caution leverage (l*) signifies the boundary of typical values for irregularities of X and it is stated as l* \(= \frac{3\left(m+1\right)}{X}\) where X stands for the sum of training compounds, and m the summation of model descriptors existent. li value > l* and standardized residual values > ± 3 within standard deviance units were seen as abnormalities of the compounds [32]. Two compounds appeared outside the warning value l* = 0.3 signifying outliers after a close observation of the AD′ (Fig. 2).
Molecular docking studies
Predicted protein–ligand relations on new investigation techniques that mix disparity change with a cavity expectation strategy were carried out using the PyRx Virtual Screen tool software [33]. Protein information bank contains Human AChEI (PDB: 4EY7) with a high-resolution 2.35 Å crystal structure. Unwanted and heteroatom particles were removed from the protein contained in the PDB record, while hydrogen was added to the protein segment and saved, the PDB was opened with Material Studio programming. The PyRx Virtual Screen tool depression location calculation and the docking was accomplished to predict the limiting method of the ligand and the unbiased protein of the scoring capacity and the saved PDB was then brought into the PyRx Virtual Screen tool interphase where the limiting pocket was guided and characterized [34].
Results
Data arrangement and QSAR models
QSAR model
The importance of the parameters selected was by virtue as the top value of R2 = 0.936, R2Adj = 0.907, Q2cv = 0.88 and R2ext = 0.881.
Descriptors elucidation
Minimum standard agreement for a reliable and influential model, the internal as well as the external justification considerations. A rise in physicochemical factors of descriptors SpMax7_Bhs, EHOMO (Homo) and MW (Molecular Weight) will increase inhibitory activities of N-aryl derivatives against Alzheimer disease since their figures were positive. Also, negative coefficients descriptors; ATS4i and MATS2e suggests that inhibitory activities of N-aryl derivatives will rise in contrast to the enzyme with decreasing standards of the descriptors (Additional file 1).
In silico design of anti-Alzheimer compound
Also, the seven designed compounds;1-(4-hydroxy-3-nitrophenyl)pyrrolidine-2,5dione (PIC50 = 1.3968),1(hydroxyphenyl)pyrrolidine-2,5-dione (PIC50 = 2.4182),1-(-4-hydroxy-3,5dinitrophenyl)pyrrlidine-2,4-dione (PIC50 = 3.124),1-(4-hydroxy-2,3,5trinitrophenyl)pyrrolidine-2,5-dione (pIC50 = 2.7643),1-(4-hydroxy-2 nitrophenyl)pyrrolidine-2,5-dione (pIC50 = 1.6064), 1-(2,4-dihydroxy-6-nitrophenyl)pyrrolidine-2,5-dione (pIC50 = 1.9319) and 1-(2,4,6-trihydroxy-3,5-dinitrophenyl)pyrrolidine-2,5-dione (pIC50 = 2.7478) were established to have better activities relative to the existing drug (Donepezil) (pIC50 = 2.7643), with compound 1-(-4-hydroxy-3,5-dinitrophenyl)pyrrlidine-2,4-dione (pIC50 = 3.124) having the largely better activity.
Molecular docking investigation and virtual screening
Investigations were carried out to regulate and calculate binding affinity towards protein target and visualize/elucidate molecular interactions between the receptor and ligand to identify types of amino acids accountable for the chemical connections at the active site of the inhibitors. Computed binding energy scores resulting from the docked complexes range from − 7.4 to − 10.1 kcal/mol with RMSD values for the majority of the complexes (over 80%) estimated to be less than 2.0 Å (Table 1). This suggests that the ligands were effectively docked to the dynamic site of the receptor and the performance of the Docking Algorithms used for this study was very reliable because a docking complex with an RMSD value less than 2 Å is considered a successful and correct docking prediction [33, 34]. The results of docked complexes with their binding energy were reported in Table 1.
ADMET property predictions and physicochemical parameters evaluation of some selected compounds
As the collaboration of an inhibitor with a compound cannot ensure its appropriateness as a medication, to further reinforce the after-effects of three-dimension-QSAR and docking studies, ADMET properties were considered on the designed molecules revealed in Table 2. The capacity to reach a target in bioactive structure was surveyed utilizing the SwissADME [35] and pkCSM [36]. Critically, the advancements carried out in these stages can envisage by a reasonable level of certainty, the false-positive outcomes ordinarily saw in biological tests of small molecules [37]. Tables 3 and 4 reveal physicochemical parameters and pharmacokinetics properties of designed compounds obtained values; Table 3 shows to be within the suitable ranges for oral bioavailability as drug candidates and obeyed Lipinski’s rule of five [38]. This implies that these compounds possessed a good physiochemical attribute generally considered in clinical trials that accounts for the good transport properties of a drug candidate [39].
Discussion
The dataset Table 5 was fragmented into 60 training set and 15 test set by clustering method. “Dt” was used in Table 5 to identify test set molecules. Several models were generated, the best was picked based on good statistical information as Eq. (4). The model 4-parametric equation containing five descriptors and an error term. Under the equation are the statistical parameters to ascertain the robustness and predictive ability of the equation. After careful validation and examination, the chosen model close by its approval boundary is introduced in Eq. (4).
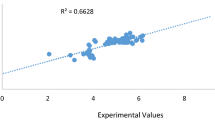
Hence, the molecule in the AD of the model can be used to make extrapolation for identified molecule without activity. Figure 1 shows the experimental activity plotted against predicted activity for training and tests sets of the model were evenly distributed on both side of the line, shown in Fig. 2, is the plot of experimental activities against the predicted activities of the test sets. The external model validation showed an exact relationship among the experimental and predicted pIC50 of the test set; also, the analytical power of the model is in the great linearity of the plot.

Experimental activity plotted against predicted activity for training and tests sets of the model

Experimental versus predicted test set value’s graph
The model William’s plot in Fig. 3 depicted their AD for the built model. A square area is bounded by 0 < l* < 0.3 and − 2.5 < SDR < 2.5, where l* is the model warning leverage in the AD of the model as described in the research.

The model William’s plot
Better action guides the application for the advancement of particles in interpreting descriptors confined in a QSAR model. Along these lines, descriptors appeared in the model of this research, their normal regression number and incidence are in Table 6. Table 7 shows the reflection on the importance of the molecular properties as well as the correlation amongst them.
Several hypothetic novel derivatives were designed using the template (Fig. 4) which was gotten from the molecule with number D35rm in the dataset (Table 5). Noble AD’s leverage value, superb standardized residual and relatively high activity are some of the factors considered in chosen the template. The facts acquired from the molecular properties confined in the model, guided the design of the derivatives. The template was modified via the adding and elimination of a selection of substituents such as –NH2, –OH, COOH and –NO2 sets. All experimental compounds (Table 5) have lesser activities compared to the seven designed compounds (Table 2).

2D structure of Ligand template
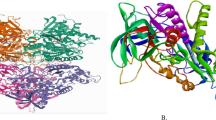
Comparison of the designed ligands and the co-crystallized ligand. Compound one (1) Van der Waals bond has THR, ASP, PHE, LEU, PHE residues which is similar to the interaction in the co-crystallized ligand, conventional Hydrogen Bond has ARG, SER, and TYR residues which is similar to the interaction in the co-crystallized ligand. Compound two (2) Van der Waals bond has LEU, ASN, SER, ARG, THR, GLN, and ILE residues which is similar to the interaction in the co-crystallized ligand, conventional Hydrogen Bond has SER, TYR, and ARG has residues which is similar to the interaction in the co-crystallized ligand, also, π–π stacked has TRP residues which is similar to the interaction in the co-crystallized ligand. Compound five (5) Van der Waals bond has VAL, LEU, THR, and GLN residues which is similar to the interaction in the co-crystallized ligand, conventional Hydrogen Bond has SER, and ARG residues which is similar to the interaction in the co-crystallized ligand and π–π Stacked has TRP residue which is similar to the interaction in the co-crystallized ligand. Compound Seven (7) Van der Waals bond has THR, PHE, and ASP residues which is similar to the interaction in the co-crystallized ligand, and conventional Hydrogen Bond has ARG, and TYR residues which is similar to the interaction in the co-crystallized ligand. Compound Eight (8) Van der Waals bond has THR, LEU, PHE, and GLN residues which is similar to the interaction in the co-crystallized ligand, conventional Hydrogen Bond has TYR, ARG, and SER residues which is similar to the interaction in the co-crystallized ligand. Compound sixteen (16) Van der Waals bond has LEU, GLN, and VAL residues which is similar to the interaction in the co-crystallized ligand, conventional Hydrogen Bond has ARG, and PHE residues which is similar to the interaction in the co-crystallized ligand. Compound Eighteen (18) Van der Waals bond has LEU, VAL, and GLN residues which is similar to the interaction in the co-crystallized ligand, conventional Hydrogen Bond has TYR, and GLN residues which is similar to the interaction in the co-crystallized ligand. All the designated compounds showed decent binding affinity and better molecular interaction at the dynamic site of the target evidenced from their binding affinity scores and the RMSD values. The binding affinity scores of the selected compounds (− 7.4 to − 10.1 kcal/mol) were found to be better compared to an FDA approved drug (Donepezil) with the binding affinity of − 6.5 kcal/mol. Also, Fig. 5 shows the structure of human AChEI (PDB ID: 4EY7), visualized through PyRx, 2D Interactions and Hydrogen bond interaction, respectively.

A Structure of human ACE (PDB ID: 4EY7), visualized through PyRx, B 2D interactions, C hydrogen bond interaction
The bioavailability radar plots showed the oral accessibility of our recommended bioactive compounds (Fig. 6), parameters of the studied molecule, make available a graphical snapshot of the drug-likeness. Lastly, Fig. 7 shows the 3D interactions of the designed compounds and the protein target.

Radar plots

3D interactions of designed ligands and protein target
Conclusions
In this work, a QSAR and virtual screening were acquired by structure-based (docking), seven ligands acquired from a blended virtual convention. As a result, we can infer that:
-
The seven chose intensifies meet these standards of no possible poisonousness, great pharmacotherapeutic profile anticipated qualities determined which is better than the reference compounds.
-
An assortment of screening techniques utilized, particularly the blended methodology, served for self-approval of the outcomes, just as to investigate, all the more effectively, the underlying variety of substance compounds, in search of a first lead.
-
The quick possibilities are purchasing these mixtures and leading broad natural movement tests (and selectivity), whose outcomes will manage the advancement of a lead is hence gotten, just as more point by point investigation of the outcomes approved.
Availability of data and materials
Not applicable.
Abbreviations
- APD:
-
Approved drug
- AD:
-
Alzheimer disease
- AD′:
-
Applicability domain
- AChEIs:
-
Acetylcholinesterase inhibitors
- ADMET:
-
Absorption Distribution Metabolism Excretion and Toxicity
- BPSD:
-
Behavioural and Psychological Symptoms of Dementia
- B3LYP:
-
Becke’s three Lee Yang and Parr
- Dt :
-
Test set
- DFT:
-
Density Functional Theory
- 2D:
-
Two dimension
- 3D:
-
Three dimension
- E (HOMO) :
-
Energy of Homo
- MF:
-
Molecular formula
- MW:
-
Molecular weight
- NRB:
-
Number of rotatable bonds
- TPSA:
-
Total polar surface area
- MR:
-
Molar refractivity
- PDB:
-
Protein Data Base
- QSAR:
-
Quantitative structure–activity relationship
- RMDS:
-
Root mean square deviation
- SDR:
-
Standardized residual
- VIF:
-
Variance inflation factor
References
Salthouse TA (2004) What and when of cognitive aging. Curr Dir Psychol Sci 13(4):140–144
Association A (2015) 2015 Alzheimer’s disease facts and figures. Alzheimer’s Dement 11(3):332–384. https://doi.org/10.1016/j.jalz.2015.02.003
Cummings JL (2000) The role of cholinergic agents in the management of behavioural disturbances in Alzheimer’s disease. Int J Neuropsychopharmacol 3(SUPPL. 2):21–29
Huang LK, Chao SP, Hu CJ (2020) Clinical trials of new drugs for Alzheimer disease. J Biomed Sci 27:1–13
Verma N, Beretvas SN, Pascual B, Masdeu JC, Markey MK (2015) New scoring methodology improves the sensitivity of the Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-Cog) in clinical trials. Alzheimer’s Res Ther 7(1):1–17
Gauthier S, Cummings J, Ballard C, Brodaty H, Grossberg G, Robert P et al (2010) Management of behavioral problems in Alzheimer’s disease. Int Psychogeriatr 22(3):346–372
Mehta D, Jackson R, Paul G, Shi J, Sabbagh M (2017) Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs 26(6):735–739. https://doi.org/10.1080/13543784.2017.1323868
Serafini MM, Catanzaro M, Rosini M, Racchi M, Lanni C (2017) Curcumin in Alzheimer’s disease: can we think to new strategies and perspectives for this molecule? Pharmacol Res 124:146–155. https://doi.org/10.1016/j.phrs.2017.08.004
Więckowska A, Kołaczkowski M, Bucki A, Godyń J, Marcinkowska M, Więckowski K et al (2016) Novel multi-target-directed ligands for Alzheimer’s disease: combining cholinesterase inhibitors and 5-HT6 receptor antagonists. Design, synthesis and biological evaluation. Eur J Med Chem 124:63–81. https://doi.org/10.1016/j.ejmech.2016.08.016
Skrzypek A, Matysiak J, Karpińska MM, Niewiadomy A (2013) Synthesis and anticholinesterase activities of novel 1,3,4-thiadiazole based compounds. J Enzyme Inhib Med Chem 28(4):816–823
Rossi E, Noberasco C, Picchi M, Di SM, Rossi A, Nurra L et al (2018) Complementary and integrative medicine to reduce adverse effects of anticancer therapy. J Altern Complement Med 24(9–10):933–941
Hung CL, Chen CC (2014) Computational approaches for drug discovery. Drug Dev Res 75(6):412–418
Macalino SJY, Gosu V, Hong S, Choi S (2015) Role of computer-aided drug design in modern drug discovery. Arch Pharm Res 38(9):1686–1701
Correa-Basurto J, Flores-Sandoval C, Marín-Cruz J, Rojo-Domínguez A, Espinoza-Fonseca LM, Trujillo-Ferrara JG (2007) Docking and quantum mechanic studies on cholinesterases and their inhibitors. Eur J Med Chem 42(1):10–19
Tong W, Hong H, Xie Q, Shi L, Fang H, Perkins R (2005) Assessing QSAR limitations—a regulatory perspective. Curr Comput Aided Drug Des 1(2):195–205
Taylor P, Ghafourian T, Cronin MTD (2015) The impact of variable selection on the modelling of oestrogenicity. SAR QSAR Environ Res 16:37–41
Duardo-sánchez A, Patlewicz G, López-díaz A (2008) Current topics on software use in medicinal chemistry: intellectual property, taxes, and regulatory issues. Curr Top Med Chem 8:1666–1675
González-Díaz H, Vilar S, Santana L, Uriarte E (2007) Medicinal chemistry and bioinformatics—current trends in drugs discovery with networks topological indices. Curr Top Med Chem 7:1015–1029
Vilar S, Cozza G, Moro S (2008) Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem 8:1555–1572
Morales A, Combes RD, Pérez M, Natália MDS (2008) Applications of 2D descriptors in drug design: a DRAGON tale. Curr Top Med Chem 8:1628–1655
Adedirin O, Uzairu A, Shallangwa GA, Abechi SE (2018) Computational studies on α-aminoacetamide derivatives with anticonvulsant activities. Beni-Suef Univ J Basic Appl Sci. 3:1–10. https://doi.org/10.1016/j.bjbas.2018.08.005
Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST et al (2006) Advances in methods and algorithms in a modern quantum chemistry program package. Phys Chem Chem Phys 8(27):3172–3191
Ambure P, Aher RB, Gajewicz A, Puzyn T, Roy K (2015) “NanoBRIDGES” software: open access tools to perform QSAR and nano-QSAR modeling. Chemom Intell Lab Syst 147:1–13. https://doi.org/10.1016/j.chemolab.2015.07.007
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inform 29(6–7):476–488
Khaled KF, Abdel-Shafi NS (2011) Quantitative structure and activity relationship modeling study of corrosion inhibitors: genetic function approximation and molecular dynamics simulation methods. Int J Electrochem Sci 6:4077–94. Available from www.electrochemsci.org
Arthur DE, Uzairu A, Mamza P, Abechi SE, Shallangwa G (2018) Insilico modelling of quantitative structure–activity relationship of pGI50 anticancer compounds on K-562 cell line. Cogent Chem 4(1):1–23. https://doi.org/10.1080/23312009.2018.1432520
Golbraikh A, Tropsha A (2002) Beware of q 2 ! J Mol Graph Modell 20:269–276
Afantitis A, Melagraki G, Sarimveis H, Igglessi-Markopoulou O, Kollias G (2009) A novel QSAR model for predicting the inhibition of CXCR3 receptor by 4-N-aryl-[1,4] diazepane ureas. Eur J Med Chem 44(2):877–884
Jaworska J, Nikolova-Jeliazkova N, Aldenberg T (2005) QSAR applicability domain estimation by projection of the training set in descriptor space: a review. Altern Lab Anim 33:445–459
Ellison CM, Sherhod R, Cronin MTD, Enoch SJ, Madden JC, Judson PN (2011) Assessment of methods to define the applicability domain of structural alert models. J Chem Inf Model 51:975–985
Tropsha A, Golbraikh A (2007) Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr Pharmaceut Des 13:3494–3504
Abdulfatai U, Uzairu A, Uba S, Shallangwa GA (2019) Molecular modelling and design of lubricant additives and their molecular dynamic simulations studies of diamond-like-carbon (DLC) and steel surface coating. Egypt J Pet 28(1):111–115. https://doi.org/10.1016/j.ejpe.2018.12.004
Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A (2020) In-silico studies of some oxadiazoles derivatives as anti-diabetic compounds. J King Saud Univ Sci 32(1):423–432
Brooijmans N (2009) Chapter: Docking methods, ligand design, and validating data sets in the structural genomics era. In: Gu J, Bourne PE (eds) Structural bioinformatics. Wiley, New York, pp 635–663
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:1–13. https://doi.org/10.1038/srep42717
Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Matlock MK, Hughes TB, Dahlin JL, Swamidass SJ (2018) Modeling small-molecule reactivity identifies promiscuous bioactive compounds. J Chem Inf Model 58(8):1483–1500
Lipinski CA (2016) Rule of five in 2015 and beyond: target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev 101:34–41. https://doi.org/10.1016/j.addr.2016.04.029
Hassan SSU, Zhang WD, Jin HZ, Basha SH, Priya SS (2022) In silico anti-inflammatory potential of guaiane dimers from Xylopia vielana targeting COX-2. J Biomol Struct Dyn 40(1):484–498. https://doi.org/10.1080/07391102.2020.1815579
Acknowledgements
The authors gratefully acknowledged the technical effort of Dr. Abdulfatai Usman, Mr Stephen Ejeh and Mr Samuel Adawara all of chemistry department, Ahmadu Bello University, Zaria.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AA designed and wrote the manuscript, UA, GAS and AES supervised and carried out the statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table 1.
Training sets and activity. Table 2. Test sets activity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ajala, A., Uzairu, A., Shallangwa, G.A. et al. 2D QSAR, design, docking study and ADMET of some N-aryl derivatives concerning inhibitory activity against Alzheimer disease. Futur J Pharm Sci 8, 30 (2022). https://doi.org/10.1186/s43094-022-00420-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-022-00420-w