
Abstract
Background
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is associated with ventricular arrhythmia, heart failure (HF), and sudden death. Thromboembolism is also an important and serious complication of ARVC/D. However, the etiology of ARVC/D and thromboembolism and their association with genetic mutations are unclear.
Methods
Genomic DNA samples of peripheral blood were conducted for whole-exome sequencing (WES) and Sanger sequencing in the ARVC/D family. Then, we performed bioinformatics analysis for genes susceptible to cardiomyopathies and arrhythmias. Further, we analyzed how the potential pathogenic mutations were affecting the hydrophobicity and phosphorylation of amino acids and their joint pathogenicity by ProtScale, NetPhos and ORVAL algorisms.
Results
We discovered a Chinese Han family of ARVC/D with right ventricular HF (RVHF), cerebral thromboembolism, arrhythmias (atrial fibrillation, atrial standstill, multifocal ventricular premature, complete right bundle block and third-degree atrioventricular block) and sudden death. Based on the WES data, the variants of LMNA p.A242V, LAMA4 p.A225P and RYR2 p.T858M are highly conserved and predicated as “deleterious” by SIFT and MetaSVM algorithms. Their CADD predicting scores are 33, 27.4 and 25.8, respectively. These variants increase the hydrophobicity of their corresponding amino acid residues and their nearby sequences by 0.378, 0.266 and 0.289, respectively. The LAMA4 and RYR2 variants lead to changes in protein phosphorylation at or near their corresponding amino acid sites. There were high risks of joint pathogenicity for cardiomyopathy among these three variants. Cosegregation analysis indicated that LMNA p.A242V might be an important risk factor for ARVC/D, electrocardiogram abnormality and cerebral thromboembolism, while LAMA4 p.A225P may be a pathogenic etiology of ARVC/D and hereditary electrocardiogram abnormality.
Conclusions
The LMNA p.A242V may participate in the pathogenesis of familial ARVC/D with RVHF and cerebral thromboembolism, while LAMA4 p.A225P may be associated with ARVC/D and hereditary electrocardiogram abnormality.
Similar content being viewed by others
What is new?
-
1.
We first demonstrated that LMNA p.A242V might participate in the pathogenesis of familial ARVC/D with right ventricular heart failure (RVHF) and cerebral thromboembolism, while LAMA4 p.A225P may be associated with ARVC/D and hereditary electrocardiogram abnormality.
-
2.
The phenotypes of LMNA p.A242V/LAMA4 p.A225P presented with ARVC/D and complex complications, including RVHF, atrial fibrillation, atrial standstill, multifocal ventricular premature, right bundle block and third-degree atrioventricular block, which were the high-risk factors for thromboembolism and sudden death.
-
3.
Anticoagulant therapy may need to be considered for patients with ARVC/D induced by LMNA p.A242V.
Introduction
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is characterized by progressive lipid storage and fibro-fatty replacement of normal cardiomyocytes in both ventricles. It is associated with ventricular arrhythmia, heart failure (HF), and sudden cardiac death (SCD), especially in young people [1,2,3]. ARVC/D commonly occurred with early precordial QRS fragmentation and malignant ventricular tachycardia of hemodynamic compromise [2, 4]. Some patients with implantable cardioverter-defibrillator (ICD) still complicate SCD induced by repeated electrical storms. Thromboembolism, especially pulmonary thromboembolism, is an important and serious complication of ARVC/D [5,6,7,8,9]. In our previous report [9], the patient with ARVC/D induced by DSG2 p.F531C mutation occurred with cerebral thromboembolism. Patients with ARVC/D and thrombus formation are at higher risk of a poor outcome and even caused sudden death induced by pulmonary thromboembolism [10]. Anticoagulation should be used in ARVC/D patients with large, hypokinetic right ventricle and slow blood flow [7]. This study found a Chinese Han family with ARVC/D, complicating bilateral low-limb edema caused by right ventricular heart failure (RVHF), complex arrhythmias, cerebral thromboembolism, hereditary electrocardiogram (ECG) abnormality and unexplained sudden death. In this ARVC/D family, four members presented with RVHF. Two of them occurred with severe cerebral thromboembolism, resulting in long-term bed rest, and one of them had sudden death at night. The etiology of ARVC/D and cerebral thromboembolism and whether thromboembolism is related to ARVC/D are unclear. Multiple genes, including desmoplakin, plakophilin-2, desmoglein-2, desmocollin-2, transforming growth factor-β3, ryanodine receptor-2 (RYR2), and transmembrane protein-43, plakoglobin, desmin, titin, phospholamban lamin A/C (LMNA), and catenin-α-3 have been identified in ARVC/D patients [11, 12]. Whole-exome sequencing (WES) combined with risk genes filter and cosegregation analysis is used to detect the possible disease-causing mutation [13]. We would further explore whether this familial ARVC/D and complex complications, especially thromboembolism, were associated with genetic background in this study.
Materials
Ethical compliance
All the participants have signed the informed consent. All procedures performed in the study involving human participants followed the Declaration of Helsinki and ethical standards approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [No.GDREC2016001H (R1)]. Detailed clinical information was collected. The clinical information included family history, age of presentation, initial symptoms of arrhythmia, physical examination, electrocardiogram (ECG), echocardiogram and cardiovascular magnetic resonance (CMR) based on their informed consent. The current study identified an ARVC/D family using the 2010 ARVC/D Revised Task Force Criteria [14].
Clinical presentation and familial characteristics
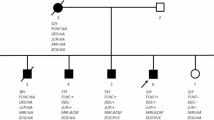
We discovered a Chinese Han family of ARVC/D (Fig. 1) with familial history of RVHF, cerebral thromboembolism, arrhythmias and unexplained sudden death. The patient of II: 4 (male, 53-year old) went to our hereditary arrhythmia clinic for genetic counseling due to a family history of cardiac edema and a complete right bundle block. He suffered from progressive right bundle block without clinical symptoms. His sister, the patient of II: 5 (female, 50-year old), occurred from atrial fibrillation for ten years and was orally administrated with Metoprolol. She felt shortness of breath when climbing two floors and appeared with severe edema of both lower extremities due to RVHF since 2016. The diagnosis of echocardiography and CMR was accordant with the primary and secondary criteria of ARVC/D. The B-type natriuretic peptide precursor of II: 5 was slightly elevated by 308.9 pg/ml. Similar to II: 5 patient, II: 1 (male, 56-year old) and II: 2 (male, 55-year old) patients also repeat complicated with obvious RVHF and severe edema of both lower extremities. The members of I: 1 (male) and II: 1 died of severer edema of low limbs induced by RVHF and cerebral ischemia stroke at the age of 50 and 56-year-old, respectively, while II: 2 occurred with severer edema of low-limbs due to RVHF and subsequently died of unexplained sudden death when sleeping at the age of 55-year old. The etiology of cerebral thromboembolism of 1: 1 and II: 1 didn’t exclude cardiogenic thromboembolism, especially when the ARVC/D pathogenesis was involved in the left ventricle. II: 4 and the third-generation family members have no arrhythmia event or clinical symptoms.

Familial pedigree
The electrocardiogram characteristics
The ECG characteristics of II: 5 present with atrial fibrillation, atrial standstill, low voltage of limb leads, multifocal ventricular premature, complete right bundle block, epsilon waves in V2-V3 lead and inverted T waves in 2016 (Fig. 2 and Table 1). The patient of II: 5 was implanted with a permanent pacemaker in another hospital due to repeat syncope induced by the third-degree atrioventricular block in 2021. The ECG of II: 3 shows the complete right bundle block, epsilon waves in V2-V4 lead and inverted T waves, while the ECG of II: 4 presents the complete right bundle block. The ECG of III: 6 indicates the early change of incomplete right bundle block, characterized by enlarging S waves in II, III, aVF, and V5-V6 leads, concomitant with abnormal q waves in III, aVF and V4-V6 leads, also flat T waves in III, V5-V6 leads and abnormal positive T wave in V1 lead. The abnormal voltage of T wave in V1 lead was higher than that of T waves in V5-V6 lead. The ECG of III: 1 shows the fat and inverted T waves in II, III, aVF and V1-V4 lead.

The electrocardiogram characteristics
The characteristics of cardiovascular magnetic resonance and echocardiogram
The CMR characteristics and echocardiogram (Fig. 3 and Table 1) of II: 5 showed obvious fat infiltration in the right ventricular free wall, decreased right ventricular wall motion, increased right ventricular volume and decreased overall systolic function, which met the main diagnostic criteria of ARVC/D. The free wall of the basal segment in the left ventricle had pericardial fat infiltration, while the left ventricular volume and the overall systolic movement were normal. There was also moderate tricuspid regurgitation.

The characteristics of cardiovascular magnetic resonance. The CMR of II: 3. a–c shows the delayed enhancement scanning PSIR images of T1W, T1W-FS and Gd-DTPA on the body axis. These images indicate the thickening of the free wall of the right ventricle with fat signal (a T1W, red arrow, and high signal; b lipid pressure of T1W, blue arrow, and low signal). Gd-DTPA delayed enhancement scanning shows mild local enhancement, suggesting the change of myocardial fibrosis. The CMR of II: 5. d–f shows the delayed enhancement scanning PSIR images of T1W, T1W-FS and Gd-DTPA on the body axis. These images indicate the thickening of the free wall of the right ventricle with fat signal (a T1W, red arrow, and high signal; b lipid pressure of T1W, blue arrow, and low signal). Figure g–i are the diastolic images of B-TFE film sequence for four-chamber heart, short-axis position and two-chamber heart of right ventricle, showing the obvious expansion of right atrium and right ventricular cavity
The CMR and echocardiogram of II: 3 indicated remarkable fat infiltration and uncoordinated movement in the right ventricular free wall, while the right ventricular systolic function was normal. Additionally, the left ventricular apex was relatively hypertrophic, and the left ventricle was slightly enlarged, although the left ventricular systolic function was normal.
The CMR of III: 6 showed normal cardiac structure and function. The echocardiograms of II: 4 and III: 6 illustrated that the middle-lower segment of the free wall in the right ventricle became thin, with some parts bulging outward like shallow sacs. The thinnest segment was in the right ventricular apex with 2.1 and 1.6 mm for II: 4 and III: 6, respectively, which also accompanied the disordered arrangement of muscular trabeculae. The echocardiogram of III: 1 showed normal cardiac structure and function by echocardiogram.
Methods
Whole exome sequencing
We extracted her blood for WES because the II: 5 proband presented a typical ARVC/D phenotype and complex complications. Genomic DNA samples of II: 5 were isolated from peripheral blood using a standard DNA extraction protocol. The isolated genomic DNA was then fragmented into 150–200 bp and subjected to DNA library preparation using established Illumina paired-end protocols. Adaptor-ligated libraries were amplified via PCR. A portion of each library was used to create an equimolar pool. Each pool was amplified to enrich targets sequenced by the Agilent SureSelectXT Target Enrichment System (Agilent Technologies Inc., Santa Clara, CA, USA). According to the manufacturer's protocol, whole-exome capture was performed with the Agilent SureSelectXT Human All Exon 50 Mb Kit (Agilent Technologies Inc.). According to the manufacturer's instructions, the exome-enriched libraries were sequenced with the Illumina Hiseq 2000 platform (Illumina, San Diego, CA, USA), and 100 bp paired-end sequencing reads were generated. Each sample was sequenced per lane to obtain an average theoretical depth of 100×.
Read mapping, variant detection, and functional annotation
After WES, raw reads were collected for quality control, in which low-quality reads were filtered, and 3′/5′ adapters were trimmed using the Trim Galore program. Clean reads were aligned to the human reference genome (University of California Santa Cruz, UCSC build hg19) using the Burrows-Wheeler Aligner (BWA) program. The quality scores were recalibrated, and reads were realigned to the reference genome using the Genome Analysis Toolkit (GATK) software package. Following the exclusion of duplicate reads, insertion-deletions (InDels) and single-nucleotide polymorphisms (SNPs) were called using the GATK or Sequence Alignment/Map tools (SAM tools).
Pathogenic risk classification
For the data from WES of II: 5 proband, SNPs and Indels were annotated using a pipeline, in which all insertion and deletion variants occurring at coding regions were considered damaging, and nonsynonymous SNPs were predicted by SIFT (http://sift.jcvi.org/www/), PolyPhen-2 (Polymorphism Phenotyping v2, http://genetics.bwh.harvard.edu/pph2/) and MetaSVM [15]. Subsequently, the common risk genes associated with cardiomyopathies and arrhythmias, as reported in our previous research [16], were detected for the II: 5 proband. These variants were screened, and the filtering criteria were as follows: (1) same variants in the WES data; (2) missense, nonsense, insertion and deletion variants; (3) SNPs with minor allele frequency, not ≥ 0.01 according to the SNP database of National Center; excluded variants with allele frequency in gnomAD (all) higher than 1%, or higher than 5% in house frequency. The potential risk variants were classified as “pathogenic (P)”, “likely pathogenic (LP)”, “uncertain significance (US/VUS)”, “likely benign (LB)” or “benign (B)” by the Clinvar database and InterVar tool [17] following the 2015 ACMG/ACP guidelines [18]. The detailed ACMG classification was shown in our previous research [16].
The variants were also excluded if the CADD predicting score was below 20. The higher CADD scores indicate that a variant is more likely to have harmful effects. A scaled score of 10 or greater indicates a raw score in the top 10% of all possible reference genome SNVs, and a score of 20 or greater indicates a raw score in the top 1%. Synonyms variants and those with a CADD predicting score lower than 20 were excluded unless reported as pathogenic or likely pathogenic in ClinVar [19].
Conservation analyses of the risk variants
To determine whether the variants, including LMNA, LAMA4 and RYR2 genes, were conserved in the species and assess their possible pathogenicity, we searched for candidate proteins from the universal protein knowledgebase (UniProt)[20]. Multiple cross-species amino acid sequences were selected in the list and extracted into FASTA format. For Multiple Sequence Alignment, T-Coffee (https://www.ebi.ac.uk/Tools/msa/tcoffee/) was used to compare the conservation of target variant sites.
Sanger sequencing in the family members
Genomic DNA samples of the family members were isolated from peripheral blood using a standard DNA extraction protocol. When the suspected pathogenic variants were obtained in each step, they were screened again using Sanger sequencing in the other family members, aiming to conduct a subsequent cosegregation analysis of genotypes and phenotypes among family members. The primers were designed by Primer Premier 5.0 and are shown in Table 2. The method of Sanger sequencing has been detailed in our previous study [11, 21]
Protein physics and chemical parameters prediction
To compare the potential functional influences of the possible risk variants on the protein's physical–chemical properties, we focused on analyzing the changes in hydrophobicity, the transmembrane domain and protein phosphorylation of amino acids induced by the mutant and wild type of LMNA, LAMA4 and RYR2 genes. ProtScale (https://web.expasy.org/protscale/) and NetPhos (http://www.cbs.dtu.dk/services/NetPhos/)were used for hydrophobicity, the transmembrane domain and phosphorylation analysis [22].
Joint pathogenicity
According to our previous study [1], LMNA, LAMA4 and RYR2 are the important risk genes participating in cardiomyopathy pathogenesis. Additionally, these three genetic variants existed in II: 5 proband. Therefore, it was necessary to analyze these three variants' joint pathogenicity and whether the joint pathogenicity aggravates the clinical phenotype of ARVC/D. ORVAL (https://orval.ibsquare.be) is the first web bioinformatics platform to explore predicted candidate disease-causing variant combinations, aiming to aid in uncovering the causes of oligogenic diseases (i.e., diseases caused by variants in a small number of genes). This tool integrates innovative machine learning methods for combinatorial variant pathogenicity prediction, further external annotations and interactive and exploratory visualization techniques [22, 23].
Results
Pathogenic risk analysis of variants
In this family (Fig. 1 and Table 3), a set of candidate genes associated with cardiomyopathies and arrhythmias were screened using the WES data of II: 5. The results showed that II: 5 carried 38 variants associated with cardiomyopathy and arrhythmia, including LMNA, ACTA1, RYR2, MYL2, MYH7, TPM1, TTN, MYOZ2, PDLIM3, LAMA4 and KIAA0196. As demonstrated in Sanger sequencing, II: 5 carried with four variants of LMNA p.A242V (NM_001257374, c.C389T), LAMA4 p.A225P (NM_001282626, c.C725T), RYR2 p.T858M (NM_001035, c.C2573T) and KIAA0196 p.H852R (NM_014846, c.A2555G). In contrast, the other variants of ACTA1, MYL2, MYH7, TPM1, TTN, MYOZ2 and PDLIM3 were negative, termed as “false positive” detected by WES.
The variants of LMNA p.A242V and LAMA4 p.A225P are predicated as “deleterious” by SIFT, Polyphen2, and MetaSVM algorithms; both of the CADD predicting scores are 33 and 27.4, respectively. According to the Clinvar database, the LMNA p.A242V (rs ID: rs397517906, gnomAD: 0.00001) has been reported in the literature in one individual affected with ARVC/D and individuals affected with DCM. This variant was also identified in three Caucasian adults (one with ARVC/D, one with clinical features of ARVC/D, and one with DCM) and segregated with disease (DCM, unspecified cardiomyopathy, chronic HF, sudden cardiac death) in six affected relatives from two families (including three obligate carriers). LMNA p.A242V was also found in the DCM family. Therefore, based on the evidence, LMNA p.A242V is classified as “LP” in the Clinvar database [https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000048076.13]. However, according to ACMG guidelines, the pathogenic risk of LMNA p.A242V is classified as “VUS”.
In contrast, the pathogenic risk of LAMA4 p.A225P (rs ID: rs782121531, gnomAD: 0.00006) is classified as “LB” and “B” by the Clinvar database and ACMG guidelines but lacked genetic verification in familial/sporadic cases of cardiovascular diseases (https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000213580.6). The variant of RYR2 p.T858M (rs ID: rs377068202, gnomAD: 0.00004) with the CADD predicting score of 25.8 is predicted as “deleterious” by the SIFT and MetaSVM algorithms but “tolerated” by the Polyphen2 algorithm. It is classified as “US” and “LB” by the Clinvar database (https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000532370.5) and ACMG guidelines due to this variant not being reported in individuals with cardiovascular disorders. The variant of KIAA0196 p.H852R [rs ID: not reported, gnomAD: not reported, according to Exome Variant Server (NHLBI Exome Sequencing Project, ESP)] with the CADD predicting score of 9.42 is predicated as “tolerated” by these three algorisms and classified as “VUS” by ACMG guidelines.
Conservation analysis
The conservation analyses (Fig. 4) demonstrate that amino acid sequences are highly conserved in the mutant sites (LMNA p.A242V and LAMA4 p.A225) among the species, including humans, mice, dogs, horses, macaque, olive baboon and zebrafish. The mutant site of RYR2 p.T858 is highly conserved among the species, including humans, mice, dogs, horses, macaque, and olive baboons, but not in the zebrafish.

The conservative analysis of the risk genetic variants
Hydrophobicity and phosphorylation prediction
The hydrophobicity analysis results (Table 4) show that the LMNA p.A242V, LAMA4 p.A225P and RYR2 p.T858M increase the hydrophobicity of their corresponding amino acid residues and their nearby sequences by 0.266, 0.378 and 0.289, respectively. The three variant sites are located in the non-transmembrane region. LMNA p.A242V does not affect the phosphorylation modification of its corresponding amino acid and nearby amino acid sequences. The LAMA4 p.A225P and RYR2 p.T858M lead to changes in protein phosphorylation at or near their corresponding amino acid sites. The LAMA4 p.A225P results in the shift of p.229Y amino acid from non-phosphorylation to phosphorylation. The RYR2 p.T858M results in the disappearance of its corresponding amino acid phosphorylation modification.
Join pathogenicity
We analyzed the joint pathogenicity of potentially candidate pathogenic sites, including the variants of LMNA p.A242V, LAMA4 p.A225P and RYR2 p.T858M. The value of support score (SS)/classification score (CS) of joint pathogenicity reached 100.00/0.95 (99% zone candidate disease-causing) for LMNA p.A242V/LAMA4 p.A225P, 100.00/0.88 (99% zone candidate disease-causing) for LAMA4 p.A225P/RYR2 p.T858M and 98.60/0.75 (95% zone candidate disease-causing) for LMNA p.A242V/RYR2 p.T858M, which indicates that there are the high risks of joint pathogenicity for cardiomyopathy among these three variants.
Cosegregation analysis
Due to III: 1 with early ECG abnormality and II: 5 proband with ARVC/D and complex complications carrying LMNA p.A242V and LMNA p.A242V/LAMA4 p.A225P (Fig. 1 and Table 5), respectively, we speculate that the patients of I: 1 and II: 1 who died of RVHF and cerebral thromboembolism also carry LMNA p.A242V. Additionally, the LAMA4 p.A225P was inherited from I: 2 without ARVC/D or ECG abnormality. These findings indicated that LMNA p.A242V might be an important risk factor for ARVC/D, ECG abnormality and cerebral thromboembolism.
The II: 3 with ARVC/D carry LAMA4 p.A225P (negative of LMNA p.A242V and RYR2 p.T858M), while the II: 4 and III: 6 with hereditary ECG abnormality carry LAMA4 p.A225P/RYR2 p.T858M, both of which inherited from I: 2, suggesting that LAMA4 p.A225P may be a pathogenic etiology of ARVC/D and hereditary ECG abnormality. The III: 7 carrying RYR2 p.T858M showed normal ECG and no ARVC/D manifestation. Therefore, we can’t determine whether RYR2 p.T858M contributes to hereditary ECG abnormality for II: 4 and III: 6 and ARVC/D for II: 5. The III: 1 with early ECG abnormality and II: 3 with ARVC/D didn’t carry RYR2 p.T858M, suggesting that the RYR2 p.T858 may not be the necessary reason for ARVC/D and ECG abnormality. For II: 2 patient, malignant ventricular arrhythmia and thromboembolism resulting from ARVC/D would lead to sudden death. Because of a lack of blood DNA samples before II: 2 death, clinical/genetic information and the possibility of incomplete penetrance, we can’t evaluate whether the RYR2 p.T858M was associated with unexplained sudden death at night.
Discussion
In this study, we first demonstrated that LMNA might participate in the pathogenesis of familial ARVC/D with RVHF and cerebral thromboembolism, while LAMA4 p.A225P may be associated with ARVC/D and hereditary ECG abnormality. The patient carrying LMNA p.A242V/LAMA4 p.A225 presented with ARVC/D and complex complications, including RVHF, atrial fibrillation, atrial standstill, multifocal ventricular premature, right bundle block and third-degree atrioventricular block, which were the high-risk factors for thromboembolism and sudden death.
LMNA p.A242V may be associated with ARVC/D and thromboembolism
LMNA, located on human chromosome 1q21.2–21.3, encodes nuclear Lamin A/C, intermediate filament proteins that are components of the nuclear laminas. The nuclear lamina is attached to the inner nuclear membrane by the binding of lamins to integral proteins of the membrane, which therefore not only functions as the structural support and mechanotransduction to the nucleus but also participates in the chromatin organization, splicing factor compartment organization, DNA replication, transcription regulation (modifying the RNA polymerase II transcription), DNA repair, posttranslational modification, signal transduction pathways and nucleocytoskeletal connections [24,25,26,27,28].
LMNA mutations abnormally increase the signaling by extracellular signal-regulated kinase 1 and kinase 2 (ERK1/2), Jun N-terminal kinase (JNK) and mitogen-activated protein kinases (MAPK), protein kinase B/mammalian target of rapamycin complex 1 and transforming growth factor-β [29,30,31]. LMNA mutations lead to a loss of nuclear stability, enhanced sensitivity to mechanical strain, drastically enhanced nuclear envelope fragility, apoptosis, endoplasmic reticulum stress, abnormal calcium handling of endoplasmic reticulum, connexin 43 downregulation and abnormal calcium oscillations upon different cellular stresses, such as hypertonic, hypoxic, oxidative stresses and mechanical stress [19, 32,33,34,35,36,37,38,39]. The inherited disorders related to LMNA mutations include muscular dystrophies, cardiac conduction defects, and cardiomyopathy (such as DCM and ARVC/D). The latter is characterized by progressive HF, complicated by life-threatening arrhythmias, and eventually death or heart transplantation [24, 40,41,42,43,44,45,46,47,48,49]. For the carriers with LMNA mutations, cardiac dysrhythmias account for 92% of patients after the age of 30-year old. HF was reported in 64% after the age of 50. Sudden death was the most frequently reported mode of death (46%) in cardiac and neuromuscular phenotypes. Carriers with LMNA mutations often received a pacemaker (28%) [50]. Moreover, there is a possible link between LMNA mutation and arterial/venous thromboembolism [40]. In our study, the LMNA p.A242V increases the hydrophobicity of its corresponding amino acid residues and their nearby sequences, which may affect the function of the LMNA protein. More interestingly, based on the cosegregation of clinical phenotypes and genotypes, our important finding first demonstrated that LMNA p.A242V might be associated with aberrant ARVC/D, severe low-limb edema induced by RVHF and thromboembolism, especially cerebral thromboembolism. Therefore, anticoagulant therapy may need to be considered for patients with ARVC/D caused by LMNA p.A242V.
LAMA4 p.A225P may be associated with ARVC/D and hereditary ECG abnormality
Laminins are important glycoproteins for the basement membrane extracellular matrix that provide cells with structural stability and signaling functions. LAMA4, the gene encoding the laminin α4 chain, is developmentally regulated during embryogenesis and localized mainly in the basement membranes of blood vessels of the adult heart and the peripheral sarcolemma of cardiomyocytes. The LAMA4 interacted with integrin molecules (especially α3ß1 integrins) and its connection with ILK. By controlling AKT kinase activity and its connection to the cytoskeleton via Parvin, the LAMA4-integrin-ILK pathway is central in converting extracellular signals into intracellular survival pathways [51].
LAMA4 knockdown in zebrafish causes defects in endothelial cells, cardiac dysfunction, and hemorrhage in embryos. LAMA4−/− mice showed microcirculation dysfunction, characterized by irregularity of capillaries and enlargement of space between cardiomyocyte and its adjacent capillary vessels, which might cause insufficient oxygen supply and cardiac phenotypes including cardiac dysfunction, cardiomyocyte degeneration and subsequent compensatory hypertrophy [52]. According to previous reports, LAMA4 p.P943L (c.2828C > T) and p.R1073X (c.3217C > T) mutations have significantly reduced the extracellular matrix in cardiomyocytes, which are associated with DCM and HCM [51]. The digenic mutations of LAMA4 p.D1309N (NM_001105206.2, c.3925G > A) and MYH7 (part of the sarcomere) p.E924K (NM_000257.3, c.2770G > A) induced infantile DCM [53]. In our study, LAMA4 p.A225P predicated as “deleterious,” increases the hydrophobicity of its corresponding amino acid residues and their nearby sequences and changes the 229th amino acid from non-phosphorylation to phosphorylation. According to the cosegregation of clinical phenotypes and genotypes, LAMA4 p.A225P may be an important pathogenic risk factor for ARVC/D and hereditary ECG abnormality.
RYR2 p.T858M is not the necessary reason for ARVC/D
The cardiac ryanodine receptor (RYR2) located in the sarcoplasmic reticulum (SR) membrane functions as a Ca2+ release channel characterized by Ca2+-induced Ca2+ release. The Ca2+ flows from the lumen of SR into the cytoplasm and activates cardiomyocyte contraction. The cardiac RYR2 protein is differentially expressed across the cardiac walls of the right ventricle, left ventricle and interventricular septum in a normal heart, with the lowest concentration expressed in the right ventricle. The RYR2 expression is reduced in all chambers in the ARVC/D canine model [54]. According to our previous study, RYR2 mutation is common in cardiac diseases, such as catecholaminergic polymorphic ventricular tachycardia, atrial fibrillation and ARVC/D, which may cause life-threatening ventricular arrhythmias and unexplained sudden death [21]. A systematic screening of the whole coding region of RYR2 in a large ARVC/D cohort without mutation in desmosome genes shows that putative RYR2 mutations are frequent (9% of ARVC/D probands) and associated with ARVC/D [55].
The pathogenic mutations (Genbank accession: No. X98330. c.5654G > A, p.G1885E and c.5656G > A, p.G1886S) of RYR2 were reported to change the subunit composition and alter the behavior of the tetramer channel complex of the RYR2 channel, which therefore causes sarcoplasmic reticulum-Ca2+ depletion and induces ARVC/D and HF [56]. A further study illustrates that the store-overload-induced calcium release activity is nearly completely abolished when mutations of p.G1885E and G1886S were introduced in the same RYR2 subunit. However, the Ca2+ loading of the intracellular stores is markedly enhanced, and the channel still displays substantial Ca2+ release on stimulation by five mM caffeine [57]. According to the cosegregation in our study, RYR2 p.T858M may not be the necessary reason for ARVC/D and ECG abnormality. Due to a lack of blood DNA samples and detailed clinical information on I: 1, II: 1 and II: 2 before death, we can’t evaluate the influence of RYR2 p.T858M on the risk of ARVC/D, thromboembolism, ECG abnormality and sudden death.
Limitation
We can’t determine the patient's genotypes of I: 1, II: 1 and II: 2 with identical phenotypes (low-limb edema induced by RVHF) due to a lack of blood DNA and detailed clinical information before death. Meanwhile, the contribution of the RYR2 variant to the risk of ARVC/D, thromboembolism, ECG abnormality and sudden death in this family cannot be determined. The joint pathogenic risks and their pathogenicity of LMNA, LAMA4 and RYR2 variants need to be further confirmed by other central data, cellular and animal experimental studies.
Conclusion
Our study first illustrated that LMNA p.A242V might participate in the pathogenesis of familial ARVC/D with RVHF and cerebral thromboembolism, while LAMA4 p.A225P may be associated with ARVC/D and hereditary ECG abnormality. Anticoagulant therapy may need to be considered for patients with ARVC/D induced by LMNA p.A242V.
Availability of data and materials
The data used in this study is not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.
Abbreviations
- ARVC/D:
-
Arrhythmogenic right ventricular cardiomyopathy/dysplasia
- HF:
-
Heart failure
- RVHF:
-
Right ventricular heart failure
- SCD:
-
Sudden cardiac death
- ICD:
-
Implantable cardioverter-defibrillator
- WES:
-
Whole-exome sequencing
- ECG:
-
Electrocardiogram
- CMR:
-
Cardiovascular magnetic resonance
- SNPs:
-
Single-nucleotide polymorphisms
- US/VUS:
-
Uncertain significance
- LB:
-
Likely benign
- B:
-
Benign
- RYR2:
-
Ryanodine receptor-2
- LMNA:
-
Phospholamban lamin A/C
- LAMA4:
-
Laminin subunit alpha 4
References
Lin Y, Zhang Q, Zhong ZA, Xu Z, He S, Rao F, et al. Whole genome sequence identified a rare homozygous pathogenic mutation of the DSG2 gene in a familial arrhythmogenic cardiomyopathy involving both ventricles. Cardiology. 2017;138(1):41–54.
Chen L, Song J, Chen X, Chen K, Ren J, Zhang N, Rao M, Hu Z, Zhang Y, Gu M, et al. A novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy provides novel insights into disease progression. Eur Heart J. 2019;40:1690–703.
Chen L, Yang F, Chen X, Rao M, Zhang NN, Chen K, Deng H, Song JP, Hu SS. Comprehensive myocardial proteogenomics profiling reveals C/EBPα as the key factor in the lipid storage of ARVC. J Proteome Res. 2017;16:2863–76.
Lin Y, Huang J, He S, Feng R, Zhong Z, Liu Y, Ye W, Li X, Liao H, Fei H, et al. Case report of familial sudden cardiac death caused by a DSG2 p.F531C mutation as genetic background when carrying with heterozygous KCNE5 p.D92E/E93X mutation. BMC Med Genet. 2018;19:148.
Gong S, Wei X, Liu G, Wu F, Chen X. Arrhythmogenic right ventricular cardiomyopathy with multiple thrombi and ventricular tachycardia of atypical left branch bundle block morphology. Int Heart J. 2018;59(3):652–4.
Hirota T, Kitaoka H, Kubo T, Okawa M, Jinnouchi Y, Furuno T, et al. Arrhythmogenic right ventricular cardiomyopathy with pulmonary embolism: a case report. J Cardiol. 2006;47(5):255–60.
Wlodarska EK, Wozniak O, Konka M, Rydlewska-Sadowska W, Biederman A, Hoffman P. Thromboembolic complications in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Europace. 2006;8(8):596–600.
Elikowski W, Błaszczyk E, Małek M, Bolewski A, Angerer D. Multiple right ventricular thrombi in arrhythmogenic right ventricular cardiomyopathy - a case report. Pol Merkur Lekarski. 2015;38(227):273–7.
Huang JN, Yang Z, Wang F, Dai YX, Ye WT, Lin YB. The thromboembolic complications associated with arrhythmogenic right ventricular cardiomyopathy/dysplasia: a case report and literature review. South China J Cardiol. 2020;21(1):64–70.
Wu L, Yao Y, Chen G, Fan X, Zheng L, Ding L, et al. Intracardiac thrombosis in patients with arrhythmogenic right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2014;25(12):1359–62.
Lin Y, Huang J, Zhao T, He S, Huang Z, Chen X, Fei H, Luo H, Liu H, Wu S, Lin X. Compound and heterozygous mutations of DSG2 identified by Whole Exome Sequencing in arrhythmogenic right ventricular cardiomyopathy/dysplasia with ventricular tachycardia. J Electrocardiol. 2018;51:837–43.
Chen L, Rao M, Chen X, Chen K, Ren J, Zhang N, Zhao Q, Yu W, Yuan B, Song J. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int J Cardiol. 2019;274:263–70.
Chen L, Zhou ZY, Lu HH, Xie Y, Li G, Huang JF, Zhao DS. Identification of a LMNA (c.646C>T) variant by whole-exome sequencing in combination with a dilated cardiomyopathy (DCM) related gene filter in a family with familiar DCM. J Biomed Res. 2018;32:314–6.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31:806–14.
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–37.
Lin Y, Huang J, Zhu Z, Zhang Z, Xian J, Yang Z, et al. Overlap phenotypes of the left ventricular noncompaction and hypertrophic cardiomyopathy with complex arrhythmias and heart failure induced by the novel truncated DSC2 mutation. Orphanet J Rare Dis. 2021;16:496.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–80.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Chirita-Emandi A, Andreescu N, Zimbru CG, Tutac P, Arghirescu S, Serban M, Puiu M. Challenges in reporting pathogenic/potentially pathogenic variants in 94 cancer predisposing genes—in pediatric patients screened with NGS panels. Sci Rep. 2020;10:223.
UniProt Consortium T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018;46:2699.
Lin Y, He S, Liao Z, Feng R, Liu R, Peng Y, Yu N, Qi H, Chen J, Huang Z, et al. Whole exome sequencing identified a pathogenic mutation in RYR2 in a Chinese family with unexplained sudden death. J Electrocardiol. 2018;51:309–15.
Zhao T, Ma Y, Zhang Z, Xian J, Geng X, Wang F, Huang J, Yang Z, Luo Y, Lin Y. Young and early-onset dilated cardiomyopathy with malignant ventricular arrhythmia and sudden cardiac death induced by the heterozygous LDB3, MYH6, and SYNE1 missense mutations. Ann Noninvasive Electrocardiol. 2021;26: e12840.
Renaux A, Papadimitriou S, Versbraegen N, et al. ORVAL: a novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res. 2019;47(W1):W93–8.
Lu JT, Muchir A, Nagy PL, Worman HJ. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech. 2011;4:562–8.
Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268:16321–6.
Crasto S, My I, Di Pasquale E. The broad spectrum of LMNA cardiac diseases: from molecular mechanisms to clinical phenotype. Front Physiol. 2020;11:761.
Zahr HC, Jaalouk DE. Exploring the crosstalk between LMNA and splicing machinery gene mutations in dilated cardiomyopathy. Front Genet. 2018;9:231.
Wang X, Zabell A, Koh W, Tang WH. Lamin A/C cardiomyopathies: current understanding and novel treatment strategies. Curr Treat Options Cardiovasc Med. 2017;19:21.
Worman HJ. Cell signaling abnormalities in cardiomyopathy caused by lamin A/C gene mutations. Biochem Soc Trans. 2018;46:37–42.
Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation. 2011;123:53–61.
Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacène E, Fromes Y, Toussaint M, Mura AM, Keller DI, et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–69.
Gerbino A, Procino G, Svelto M, Carmosino M. Role of lamin A/C gene mutations in the signaling defects leading to cardiomyopathies. Front Physiol. 2018;9:1356.
Brayson D, Shanahan CM. Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus. 2017;8:17–33.
Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet. 2013;22:2335–49.
Davidson PM, Lammerding J. Broken nuclei–lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014;24:247–56.
Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2005;14:2167–80.
Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–20.
Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Investig. 2004;113:370–8.
Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Investig. 2004;113:357–69.
van Rijsingen IA, Bakker A, Azim D, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Christiaans I, Lekanne Dit Deprez RH, Wilde AA, et al. Lamin A/C mutation is independently associated with an increased risk of arterial and venous thromboembolic complications. Int J Cardiol. 2013;168:472–7.
Chen SN, Sbaizero O, Taylor M, Mestroni L. Lamin A/C cardiomyopathy: implications for treatment. Curr Cardiol Rep. 2019;21:160.
Captur G, Arbustini E, Bonne G, Syrris P, Mills K, Wahbi K, Mohiddin SA, McKenna WJ, Pettit S, Ho CY, et al. Lamin and the heart. Heart. 2018;104:468–79.
Goidescu CM. Dilated cardiomyopathy produced by lamin A/C gene mutations. Clujul Med. 2013;86:309–12.
Kato K, Takahashi N, Fujii Y, Umehara A, Nishiuchi S, Makiyama T, Ohno S, Horie M. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J Cardiol. 2016;68:346–51.
Cattin ME, Muchir A, Bonne G. “State-of-the-heart” of cardiac laminopathies. Curr Opin Cardiol. 2013;28:297–304.
Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715–26.
Fu Y, Eisen HJ. Genetics of dilated cardiomyopathy. Curr Cardiol Rep. 2018;20:121.
Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–80.
Liang JJ, Grogan M, Ackerman MJ, Goodsell K. LMNA-mediated arrhythmogenic right ventricular cardiomyopathy and Charcot-Marie-Tooth Type 2B1: a patient-discovered unifying diagnosis. J Cardiovasc Electrophysiol. 2016;27:868–71.
van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, de Visser M, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death. J Mol Med. 2005;83:79–83.
Knöll R, Postel R, Wang J, Krätzner R, Hennecke G, Vacaru AM, Vakeel P, Schubert C, Murthy K, Rana BK, et al. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation. 2007;116:515–25.
Wang J, Hoshijima M, Lam J, Zhou Z, Jokiel A, Dalton ND, Hultenby K, Ruiz-Lozano P, Ross J Jr, Tryggvason K, et al. Cardiomyopathy associated with microcirculation dysfunction in laminin alpha4 chain-deficient mice. J Biol Chem. 2006;281:213–20.
Abdallah AM, Carlus SJ, Al-Mazroea AH, Alluqmani M, Almohammadi Y, Bhuiyan ZA, Al-Harbi KM. Digenic inheritance of LAMA4 and MYH7 mutations in patient with infantile dilated cardiomyopathy. Medicina (Kaunas). 2019;55:17.
Meurs KM, Lacombe VA, Dryburgh K, Fox PR, Reiser PR, Kittleson MD. Differential expression of the cardiac ryanodine receptor in normal and arrhythmogenic right ventricular cardiomyopathy canine hearts. Hum Genet. 2006;120:111–8.
Roux-Buisson N, Gandjbakhch E, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Mansencal N, Delacretaz E, Cosnay P, et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: results of a systematic screening. Heart Rhythm. 2014;11:1999–2009.
Milting H, Lukas N, Klauke B, Körfer R, Perrot A, Osterziel KJ, Vogt J, Peters S, Thieleczek R, Varsányi M. Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Res. 2006;71:496–505.
Koop A, Goldmann P, Chen SR, Thieleczek R, Varsányi M. ARVC-related mutations in divergent region 3 alter functional properties of the cardiac ryanodine receptor. Biophys J. 2008;94:4668–77.
Acknowledgements
We thank Shulin Wu, Chunyu Deng, Zhixin Shan and Fang Rao from Guangdong Cardiovascular Institute. We are grateful to Jinsheng Tao, BGI (Shenzhen, China), for technical assistance.
Funding
This work was supported by the Science and Technology Project of Zhuhai [20191210E030072], the Science Project of the Second People's Hospital of Guangdong Province [TQ2019-005] and the Medical Science and Technology Research Project of Guangdong Province [A2020069].
Author information
Authors and Affiliations
Contributions
JC, HL and ZL: pedigree analysis and writing; YTM: bioinformatics analysis; HL: Sanger sequencing; YPL, ZY, FW and QZ: imaging analysis, case collection, and follow up; ZBY and YBL: quality control of clinical data and clinical design. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were following the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The protocol was approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [No.GDREC2016001H (R1)].
Consent for publication
Not applicable.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Competing interests
The authors of this study declare that they each have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, J., Ma, Y., Li, H. et al. Rare and potential pathogenic mutations of LMNA and LAMA4 associated with familial arrhythmogenic right ventricular cardiomyopathy/dysplasia with right ventricular heart failure, cerebral thromboembolism and hereditary electrocardiogram abnormality. Orphanet J Rare Dis 17, 183 (2022). https://doi.org/10.1186/s13023-022-02348-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02348-z