
Abstract
The hazel dormouse Muscardinus avellanarius presents an exemplary non-model species that is both locally threatened and whose genetic status is not fully understood owing to insufficient resolution of the currently available molecular tools. We performed normalized Genotyping-by-Sequencing (nGBS) on 48 hazel dormouse samples collected across the species European distribution, aiming at discovering useful single nucleotide polymorphism (SNP) markers for the assessment of population structure and genomic diversity. The analyses of > 24,000 SNPs showed a high divergence between the Eastern and Western lineage of the species with high rates of SNP allele fixation, consistent with previous studies suggesting the divergence of lineages occurred over 2 mya. These results indicate that investigating inter-lineage as well as within-lineage genetic composition will be a conclusive approach for identifying conservation strategies in the future. Results presented here indicate the highest genetic divergence in the Italian and Lithuanian populations. We document how nGBS allows the discovery of SNPs that can characterize patterns of genetic variation at multiple spatial scales in a non-model organism. We document how nGBS allows the discovery of SNPs that can characterize patterns of genetic variation at multiple spatial scales in a non-model organism, potentially informing monitoring and conservation strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The hazel dormouse (Muscardinus avellanarius, linnaeus, 1758) is currently undergoing population decline in many areas of its European distribution. Possible reasons are habitat fragmentation, decreasing habitat quality and climate change (Bright et al. 2006; Goodwin et al. 2018, Mortelliti et al. 2010, 2014). Despite being internationally protected and the subject of several ongoing conservation projects, hazel dormouse populations are still decreasing annually in parts of Europe (Goodwin et al. 2017; Schulz and Büchner 2018; Hutterer et al. 2021). For instance, populations in the north-western range have been reduced by up to 72% in the last decades (Goodwin et al. 2017). In addition to this sharp decline, knowledge regarding some genetic aspects of the biology of this arboreal species is still incomplete (Mouton et al. 2017).
In particular, hazel dormouse research has not yet benefitted from the wide range of genomic techniques now available to study non-model organisms (Allendorf et al. 2010; Funk et al. 2012). Advances in molecular genetic techniques have made next generation sequencing (NGS) a staple in conservation and population genetics studies (DeSalle and Amato 2004; Allendorf et al. 2010). NGS allows for a higher resolution of population genetic parameters, such as effective population size, inbreeding, population structure,which may not be possible with traditional marker sets (Hohenlohe et al. 2021). The hazel dormouse is a candidate species to benefit from NGS as this species is largely arboreal, nocturnal and generally remains at low population densities, making it challenging to detect and logistically prohibitive to monitor on a large scale (Goodwin et al. 2017). Given these constraints, genetic solutions can inform applied conservation strategies for this elusive rodent.
Studies based on mitochondrial DNA (mtDNA) analysis and autosomal microsatellites have suggested that the hazel dormouse is split into two genetically distinct lineages or even cryptic species (Western lineage and Eastern lineage) in Europe (Mouton et al. 2012a, b, 2017). Further, Bani et al. (2017) used eight microsatellites to assess the influence of habitat fragmentation on the genetic diversity in the hazel dormouse and found inbreeding and genetic drift to occur in populations living in fragmented landscapes. Such genetic factors can play an important role in the health and viability of populations occurring in small, isolated patches (Frankham 2005). These initial genetic results indicate that the hazel dormouse is highly divergent, both at a large scale, given the presence of two distinct lineages, and at a fine-scale, due to habitat fragmentation and limited dispersal capability. Consequently, a rigorous assessment of genetic substructuring may reveal evolutionary significant units that are extremely valuable for future conservation strategies.
In this study, we aimed to leverage nGBS technologies on hazel dormouse populations to (i) examine the present genetic structuring among populations and (ii) discover informative SNP markers at both broad and fine-scale. We provide a foundation for further genomic analyses on hazel dormouse populations, which will be able to be used for future developments in scientific assessment, monitoring and conservation, especially in regions where populations are currently declining.
Methods
Sample preparation
We obtained 98 tissue samples from across the European distribution of the species, which were collected between 2008 and 2018 (Supplementary Table S1). Of the collected samples, 74 originated from the known range of the Eastern lineage (EL) and 24 were individuals from the Western lineage (WL) (Mouton et al. 2017), representing both main lineages of the hazel dormouse in continental Europe. We divided the samples into 16 populations classified solely based on geographic sampling locations (Supplementary Table S1).
DNA was extracted using the Qiagen DNeasy Blood & Tissue Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol with the following slight modifications: (i) to receive RNA-free genomic DNA, 4 µL of RNAse A were added during the lysis step at 56 °C, (ii) after the washing steps the spin columns were centrifuged for 3 min at 13,300xg and dried at 56 °C for 5–10 min, (iii) to increase the yield, DNA was eluted in two steps with 40 µL AE buffer each. After incubation for 5 min at room temperature, the columns were centrifuged at 13,000xg for 1 min resulting in a total DNA extract volume of 80 µL per sample. Samples were only sent for nGBS if ≥ 300 ng of high molecular weight DNA could be provided, as specified by the protocol from Biosearch Technologies Genomic Analysis by LGC (www.biosearchtech.com) (Arvidsson et al. 2016).
Sequencing protocol
Library preparation and sequencing was performed according to the LGC protocol for nGBS (Arvidsson et al. 2016). Briefly, genomic DNA was digested using the restriction enzyme MslI. Next, the restriction digests were mixed with one of 96 inline-barcoded forward blunt adaptors, followed by ligation and thus converted into GBS libraries. The DNA libraries were then purified using magnetic beads. After purification, DNA was PCR-amplified using standard Illumina TrueSeq amplification primers. The normalization step was performed using Trimmer Kit (Evrogen), which reduces the number of abundant fragments, and subsequently reamplified again using 14 PCR cycles. The nGBS libraries were then size selected on Blue Pippin, discarding fragments smaller than 300 bp and larger than 500 bp long. Sequencing was carried out on an Illumina NextSeq 500/550 using V2 Chemistry and 300 cycles. Sequences were demultiplexed with Illumina’s CASAVA data analysis software and proprietary software developed by LGC Genomics.
Upon receiving the sequencing data, the demultiplexed reads were first trimmed to remove adapter sequences as well as stretches of low quality and ambiguous bases using Trimmomatic v.0.38 (Bolger et al. 2014). Then, the reads were mapped to the hazel dormouse reference genome (PRJNA399340, Broad Institute) using BWA-MEM (v 0.7.12-r1039) with default parameters (Li und Durbin 2009). SNP calling was carried out using samtools (v1.9) (Li et al. 2009; Li 2011) mpileup function. The called raw SNPs were filtered out if loci showed > 15% missing data across samples, minor allele frequency (MAF) was lower than 0.05, sequencing depth was lower than 3.0 x or higher than twice the mean depth, or if loci showed a genotype quality < 20. In addition to these criteria, SNPs were also pruned to account for linkage disequilibrium using r2 > 0.5 in 100 kb windows using bcftools (v1.9) (Li et al. 2009).
Population genomic analyses
To understand structuring and to have first trends in allele frequencies within and among the sampled populations, we first performed a principal component analysis (PCA) using PLINK (v1.9) (Chang et al. 2015), which utilizes a variance-standardized relationship matrix which requires no a priori information on populations. We then calculated genotype likelihoods using ANGSD (v0.930) (Korneliussen et al. 2014). Here, we utilized the samtools model to estimate likelihoods with a SNP p-value of 1e−6. The software ADMIXTURE (v 1.3.0) (Alexander and Lange 2011) was subsequently used to infer population structure between individuals using K = 1 to K = 20 with 50 repetitions of each K allowing for investigation of convergence patterns. We merged the resulting runs using CLUMPAK (Kopelman et al. 2015) and determined the optimal K based on the Evanno method (Evanno et al. 2005). To investigate population differentiation, we calculated population pairwise FST values using ANGSD. Using a two-dimensional site frequency spectrum (2dSFS) for each pair of populations and the reference genome as the ancestral genotype, we calculated the weighted FST estimates. Lastly, we performed calculations of genomic diversity for each of the 16 geographically identified populations using the population function in STACKS (v.2.41) (Catchen et al. 2013). The genomic diversity values for the EL and WL were calculated separately to prevent underestimation of multiple values due to fixation of a high number of alleles in each of the major lineages.
Results
DNA extraction and sequencing
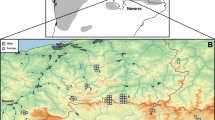
DNA extraction resulted in 48 samples (49%) meeting the quality specifications for nGBS, i.e. the required quantity and quality of DNA. After sequencing and demultiplexing at LGC, we received on average 3 million of reads per individual with an average coverage of 15.08 x. Mapping to the hazel dormouse reference genome resulted in an average alignment rate of 99.5%. Three samples, from German (n = 2) and Czech (n = 1) populations, mapped below 60% to the reference genome and were subsequently removed from downstream analysis. Further, one German and one Belgian sample could not be assigned to exact spatial coordinates and were excluded from further analysis. After SNP filtering, we identified 24,903 polymorphic SNP loci across the genome in 43 individuals representing seven populations (Fig. 1; Italy, Belgium, Netherlands, Baden-Wuerttemberg, Hesse, Saxony, and Lithuania; see Supplementary Table S1 for more details). The average depth across individuals was 11.9 x after filtering, with a range spanning from 3.9 x to 24.3 x.

Sample map showing the 43 hazel dormouse individuals that were successfully sequenced and used for downstream analysis. The shaded grey marks the known hazel dormouse distribution according to the Hutterer et al. (2016). Colors indicate the most likely K identified through ADMIXTURE analysis. The dashed line indicates the fictive contact zone between the Western and Eastern lineage as proposed by Mouton et al. (2017)
Population genomic analyses
The resulting 24,903 SNPs were used to perform basic statistical analyses on population differentiation and genetic diversity. The PCA analysis identified two main clusters, which coincided with the expected split between the EL and WL. In total, 91.5% of the variance in identified SNPs is explained through this axis (Fig. 2A). An additional separation was found between the Lithuanian and the German populations (Hesse, Saxony, Baden-Wuerttemberg) (Fig. 2A). The ADMIXTURE analysis revealed K = 3 clusters as most likely, which equally confirmed the split between the lineages, and the Lithuanian population as a separate third cluster. Higher values of K showed the separation of the Italian populations and some substructuring within the German populations.

PCA results from 43 hazel dormouse individuals and 24,903 SNPs. Blue colored symbols represent populations from the Eastern lineage (BW = Baden-Wuerttemberg, HE = Hesse, LT = Lithuania, SN = Saxony-Anhalt) and orange-colored symbols represent populations from the Western lineage (BE = Belgium, IT = Italy, NL = Netherlands). The different plots show results for A All individuals, B 29 individuals from the Eastern lineage, and C 14 individuals from the Western lineage
PCA analysis carried out on each lineage separately confirmed the split between the Lithuanian and all German populations on the first axis (42.6% variance explained). In addition, German populations showed some differentiation, with a gradient from Hesse to Saxony (Figs. 2B and 8.36% variance explained). The population in Saxony showed a high degree of within population variance. In the WL, the Italian population is separated from the Belgian and Dutch populations (39.8% variance explained) (Fig. 2C). Pairwise FST values confirmed these results, which identified the Italian population as divergent within the WL (Supplementary Figure S1).
Observed heterozygosity (Ho) values for each population ranged from 0.17 to 0.29 (Supplementary Table S2). WL individuals showed on average higher heterozygosity values compared to EL (Ho = 0.26 for WL, and 0.18 for EL, respectively). Private alleles calculated across all lineages were found in all populations; however, the number varied greatly ranging from five in Baden-Wuerttemberg to 586 in Lithuania. Specifically, the Lithuanian, Hesse, and Italian populations showed the greatest number of private alleles (Supplementary Table S2).
Discussion
In this study, 48 dormouse individuals from seven populations were sequenced and 24,903 SNP loci were identified, which were successfully used to study lineage differentiation, population structure and genomic diversity. These are promising for future use in conservation management of this species. The first aim of this study was to investigate the genetic structure at both a broad and fine-scale. In this context, we found a strong divergence between the EL and WL, in line with previous findings, which evidenced that the divergence between the two lineages likely occurred over 2.76 mya (Mouton et al. 2017). Such a long history of divergence may explain our finding of a substantial degree of SNPs being fixed among the lineages. While these loci are useful for lineage discrimination, they can cover weaker signals of population substructuring occurring within lineages and populations.
At a more regional scale, all populations within the EL showed moderate levels of observed heterozygosity. Interestingly, there is a higher degree of within population diversity in the Saxony population. These results indicate the presence of genetic differentiation on a regional scale, as shown by earlier work based on microsatellite markers (Bani et al. 2017). This trend stems from the species’ limited dispersal capability, which is further diminished through habitat fragmentation (Bani et al. 2017). The higher number of private alleles identified in some populations (Supplementary Table S2) may be an indication of genetic differentiation. However, a more extensive, evenly distributed sampling strategy would be needed to verify this. Further, the genetic differentiation in Italy confirms the knowledge regarding the isolation of populations during the Last Glacial Maximum, where Italy served as a refuge leaving unique genomic signatures across a variety of taxa (Hewitt 1999, 2000).
The second aim was to identify informative SNP markers for further use in conservation efforts for this endangered rodent. We detected a large pool of SNPs that can be utilized for future studies on the hazel dormouse. Specifically, studies looking at fine-scale patterns of genetic diversity and differentiation, as well as range-wide phylogeography of the species will add valuable knowledge about the demographic history of this species and will contribute to the identification of evolutionary significant units important for conservation. Previous studies have documented the applicability of reduced SNP panels to detect regional population structures and cryptic species (Antao et al. 2011; Muñoz et al. 2015) and to allow for a high-resolution genetic monitoring of threatened species (Kraus et al. 2015; von Thaden et al. 2017). Adapting such a panel for a platform enabling accurate genotyping of noninvasively collected samples (von Thaden et al. 2017, 2020), such as hairs or scats, will greatly improve its applicability in monitoring and conservation projects for this small, elusive and protected mammal.
References
Alexander DH, Lange K (2011) Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform 12(1):246. https://doi.org/10.1186/1471-2105-12-246
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11(10):697–709. https://doi.org/10.1038/nrg2844
Antao T, Pérez-Figueroa A, Luikart G (2011) Early detection of population declines: high power of genetic monitoring using effective population size estimators. Evol Appl 4(1):144–154. https://doi.org/10.1111/j.1752-4571.2010.00150.x
Arvidsson S, Fartmann B, Winkler S, Zimmermann W (2016) : Efficient high-throughput SNP discovery and genotyping using normalised Genotyping-by-Sequencing (nGBS). Edited by LGC Limited. Berlin, Germany (AN-161104.01)
Bani L, Orioli V, Pisa G, Fagiani S, Dondina O, Fabbri E et al (2017) Population genetic structure and sex-biased dispersal of the hazel dormouse (Muscardinus avellanarius) in a continuous and in a fragmented landscape in central Italy. Conserv Genet 18(2):261–274. https://doi.org/10.1007/s10592-016-0898-2
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford) 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bright PW, Morris PA, Mitchell-Jones T (2006) The Dormouse conservation handbook, 2nd edn. English Nature, Petersborough
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22(11):3124–3140. https://doi.org/10.1111/mec.12354
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4:7. https://doi.org/10.1186/s13742-015-0047-8
DeSalle R, Amato G (2004) The expansion of conservation genetics. Nat Rev Genet 5(9):702–712. https://doi.org/10.1038/nrg1425
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Frankham R (2005) Stress and adaptation in conservation genetics. J Evol Biol 18(4):750–755. https://doi.org/10.1111/j.1420-9101.2005.00885.x
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27(9):489–496. https://doi.org/10.1016/j.tree.2012.05.012
Goodwin CED, Hodgson DJ, Al-Fulaij N, Bailey S, Langton S, McDonald RA (2017) Voluntary recording scheme reveals ongoing decline in the United Kingdom hazel dormouse Muscardinus avellanarius population. Mam Rev 47(3):183–197. https://doi.org/10.1111/mam.12091
Goodwin CED, Suggitt AJ, Bennie J, Silk MJ, Duffy JP, Al-Fulaij N, Bailey S, Hodgson DJ, McDonald RA (2018) Climate, landscape, habitat, and woodland management associations with hazel dormouse Muscardinus avellanarius population status. Mam Rev 48:209–223
Hewitt G (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68(1–2):87–112. https://doi.org/10.1111/j.1095-8312.1999.tb01160.x
Hewitt G (2000) The genetic legacy of the quaternary ice ages. Nature 405(6789):907–913. https://doi.org/10.1038/35016000
Hohenlohe PA, Funk WC, Rajora OP (2021) Population genomics for wildlife conservation and management. Mol Ecol 30(1):62–82. https://doi.org/10.1111/mec.15720
Hutterer R, Kryštufek B, Yigit N, Mitsain G, Meinig H, Juškaitis R (2016) The IUCN red list of threatened species. https://doi.org/10.2305/IUCN.UK.2016-3.RLTS.T13992A22222242.en
Hutterer R, Kryštufek B, Yigit N, Mitsainas G, Meinig H, Juškaitis R (2021) Muscardinus avellanarius (amended version of 2016 assessment). The IUCN Red List of Threatened Species 2021: e.T13992A197519168. https://doi.org/10.2305/IUCN.UK.2021-1.RLTS.T13992A197519168.en. Accessed 13 December 2021
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res 15(5):1179–1191. https://doi.org/10.1111/1755-0998.12387
Korneliussen TS, Albrechtsen A, Nielsen RANGSD (2014) Analysis of next generation sequencing data. BMC Bioinform 15:356. https://doi.org/10.1186/s12859-014-0356-4
Kraus RHS, vonHoldt B, Cocchiararo B, Harms V, Bayerl H, Kühn R et al (2015) A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Res 15(2):295–305. https://doi.org/10.1111/1755-0998.12307
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics (Oxford) 27(21):2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics (Oxford) 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics (Oxford) 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Mortelliti A, Amori G, Capizzi D, Rondinini C, Boitani L (2010) Experimental design and taxonomic scope of fragmentation studies on European mammals: current status and future priorities. Mammal Rev 40(2):125–154. https://doi.org/10.1111/j.1365-2907.2009.00157.x
Mortelliti A, Sozio G, Driscoll DA, Bani L, Boitani L, Lindenmayer DB (2014) Population and individual-scale responses to patch size, isolation and quality in the hazel dormouse. Ecosphere 5(9):art107. https://doi.org/10.1890/ES14-00115.1
Mouton A, Grill A, Maurizio S, Kryštufek B, Randi E, Amori G et al (2012) Evidence of a complex phylogeographic structure in the common dormouse, Muscardinus avellanarius (Rodentia: Gliridae). Biol J Linn Soc 105(3):648–664. https://doi.org/10.1111/j.1095-8312.2011.01807.x
Mouton A, Grill A, Maurizio S, Kryštufek B, Randi E, Amori G et al (2012) Using phylogeography to promote dormouse conservation: the case of Muscardinus avellanarius (Rodentia, Gliridae). Peckiana 8:255–264
Mouton A, Mortelliti A, Grill A, Sara M, Kryštufek B, Juškaitis R et al (2017) Evolutionary history and species delimitations: a case study of the hazel dormouse, Muscardinus avellanarius. Conserv Genet 18(1):181–196. https://doi.org/10.1007/s10592-016-0892-8
Muñoz I, Henriques D, Johnston JS, Chávez-Galarza J, Kryger P, Pinto MA (2015) Reduced SNP panels for genetic identification and introgression analysis in the dark honey bee (Apis mellifera mellifera). PLoS ONE 10(4):e0124365. https://doi.org/10.1371/journal.pone.0124365
Schulz B, Buchner S (2018) Populations of Muscardinus avellanarius in north-western Europe can survive in forest poor landscapes, when there are enough hedges (Rodentia: Gliridae). Lynx Series Nova 49:57–68
von Thaden A, Cocchiararo B, Jarausch A, Jüngling H, Karamanlidis AA, Tiesmeyer A et al (2017) Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Sci Rep 7(1):10768. https://doi.org/10.1038/s41598-017-10647-w
von Thaden A, Nowak C, Tiesmeyer A, Reiners TE, Alves PC, Lyons LA et al (2020) Applying genomic data in wildlife monitoring: development guidelines for genotyping degraded samples with reduced single nucleotide polymorphism (SNP) panels. Mol Ecol Res. https://doi.org/10.1111/1755-0998.13136
Acknowledgements
This study was partially funded by the Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz of the German Federal State of Hesse (LOEWE). Part of funding for sample collection came from the DFG Grant DFG: FI 831/6-1 (Joanna Fietz).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leyhausen, J., Cocchiararo, B., Nowak, C. et al. Genotyping-by-sequencing based SNP discovery in a non-model rodent, the endangered hazel dormouse. Conservation Genet Resour 14, 195–201 (2022). https://doi.org/10.1007/s12686-022-01253-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-022-01253-8