
Abstract
Bacterial spot (BS), caused by Xanthomonas euvesicatoria, X. vesicatoria, X. gardneri and X. perforans, is an economically important bacterial disease of tomato and pepper. Symptoms produced by all four species are nearly indistinguishable. At present, no point-of-care diagnostics exist for BS. In this research, we examined genomes of X. euvesicatoria, X. vesicatoria, X. gardneri, X. perforans and other species of Xanthomonas; the unique gene recG was chosen to design primers to develop a loop-mediated isothermal amplification (LAMP) assay to rapidly and accurately identify and differentiate X. euvesicatoria from other BS causing Xanthomonas sp. using a field-deployable portable BioRangerTM instrument. Specificity of the developed assay was tested against 39 strains of X. euvesicatoria and 41 strains of other species in inclusivity and exclusivity panels, respectively. The assay detection limit was 100 fg (~18 genome copies) of genomic DNA and 1,000 fg in samples spiked with tomato DNA. The assay unambiguously detected X. euvesicatoria in infected tomato plant samples. Concordant results were obtained when multiple operators performed the test independently. No false positives and false negatives were detected. The developed LAMP assay has numerous applications in diagnostics, biosecurity and disease management.
Similar content being viewed by others

Introduction
Bacterial spot (BS) of tomato (Solanum lycopersicum) and pepper (Capsicum spp.) is one of the most serious and economically important diseases worldwide. The disease is caused by four species of Xanthomonas, Xanthomonas euvesicatoria, X. perforans, X. vesicatoria and X. gardneri1. This disease can reduce the yield up to 50%2,3. Warm and humid climates favor disease development on tomato and pepper, which are both susceptible to X. euvesicatoria, X. vesicatoria and X. gardneri (cluster in group A, B and D, respectively), while the pathogenicity of X. perforans (group C) is limited to tomato2,4. In the beginning of the disease development water soaked lesions on the upper and lower epidermis of leaves can be observed. Additionally, bacterial spot symptoms on tomato and pepper include on leaves and fruits, defoliation and spotting on the stem; but the leaf symptoms fluctuate based on environmental conditions3,5. The pathogen X. euvesicatoria is widely distributed throughout the world6, but symptoms produced by X. euvesicatoria, X. vesicatoria, X. gardneri and X. perforans cannot be distinguished in field settings. Therefore, new tools are required to precisely and rapidly identify X. euvesicatoria for accurate and timely management of the disease.
The accurate and timely detection of plant pathogens is not only a critical criteria for disease management but also for regulatory issues7,8. Plant pathogenic xanthomonads can be identified based on carbon utilization patterns and fatty acid profiles, but DNA based technologies have been more popular recently9 because of their high specificity and sensitivity7. Currently, xanthomonads are identified using Multilocus Sequence Typing (MLST), Amplified Fragment Length Polymorphism (AFLP), DNA-DNA hybridization, and other polymerase chain reaction-based methods including end-point PCR, multiplex PCR, and real-time quantitative PCR (qPCR)10,11,12. The four Xanthomonas species, which cause bacterial spot of tomato and pepper were differentiated using DNA-DNA hybridization1.
Several species-specific PCR and qPCR assays have been developed for the specific detection of X. euvesicatoria, X. vesicatoria and X. gardneri and X. perforans9,13,14,15. However, PCR and qPCR based methods require sophisticated and expensive equipment, and usually cannot be performed at point-of-care. Recent advances in isothermal amplification methods have the ability to rapidly identify pathogens with minimal laboratory equipment; results can be obtained within 10 minutes. Isothermal amplification reactions are performed at a constant temperature and are usually more tolerant to inhibitors compared to PCR and qPCR16. Currently, there are several different types of isothermal methods available including, recombinase polymerase amplification (RPA)17, strand displacement amplification (SDA)18, helicase-dependent amplification (HDA)19, nicking enzyme amplification reaction (NEAR)20, loop-mediated isothermal amplification (LAMP)21, and rolling circle amplification (RCA)22,23. LAMP is the most popular and widely used isothermal-based detection method because its rapid, ease to perform and also has greater sensitivity and is compatible with numerous detection chemistries. Most importantly, it can be easily performed at the point-of-care. It has been successfully used for rapid and specific detection of plant bacteria from infected plant tissues and soil24,25.
LAMP utilizes a strand displacing, DNA polymerase, a set of two inner [hybrid] primers (FIP and BIP) and two outer primers (F3 and B3)26. The reaction is initiated by the inner primer (either FIP or BIP) hybridizing to its priming site (F2c or B2c) on the target DNA. The outer primer (F3 or B3) secondarily hybridizes to its priming site (F3c or B3c) on the target DNA and initiates synthesis of a new complementary sequence that displaces the DNA sequences extended from the inner primer. The outcome is a DNA sequence which can form stem-loop structures at both ends27,28. Inclusion of internal loop primers (LF and LB) accelerate the LAMP reaction and further reduce the total reaction time27. The visualization of the amplification products can be obtained using several methods including gel electrophoresis, measuring turbidity and visually evaluating the color change by SYBR Green stain.
The objective of this study was to develop a point-of-care LAMP protocol for specific and rapid detection of X. euvesicatoria from purified, mixed cultures and infected plant tissues. These developed protocols have applications in plant pathology for routine diagnostics, surveillance, biosecurity, epidemiology and disease management.
Results
Genome comparison, primer design and in silico validation
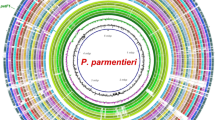
Comparison of 10 genomes of the genera Xanthomonas, Dickeya, Pectobacterium and Ralstonia allowed the unique gene selection for development of a specific LAMP assay for X. euvesicatoria. The genomes were evaluated using two approaches, BLAST comparison and OrthoANI (average nucleotide identity) (Fig. 1). Xanthomonas species sharing a high genome similarity were grouped together (Fig. 1A and B). Regardless of causing similar symptoms on the same hosts, X. euvesicatoria, X. vesicatoria, X. gardneri and X. perforans were clustered in two subgroups (Fig. 1B). X. euvesicatoria and X. perforans genomes showed highest similarity of 98.5% within the BS-causing Xanthomonas species. However, X. vesicatoria and X. gardneri shared 86.5% ANI similarity and were grouped together (Fig. 1B). Dickeya, Pectobacterium and Ralstonia showed less than 70% similarity with any of the Xanthomonas species and were grouped outside.

Target gene selection and genomic variation. (A) A ring image was generated to locate the recG gene region. Genomes of Xanthomonas euvesicatoria (NZ_CP018467), X. vesicatoria (NZ_CP018725), X. gardneri (NZ_CP018731), X. perforans (NZ_CP019725), X. campestris pv. campestris (NZ_CP012145), Dickeya solani (NZ_CP015137), X. axonopodis pv. glycines (NZ_CP017188), X. axonopodis pv. dieffenbachiae (NZ_CP014347), Pectobacterium carotovorum subsp. carotovorum (NC_018525) and Ralstonia solanacearum (NC 003295) were retrieved from NCBI GenBank genome database. In mapped genome ring image from the inside out shows: genome coordinates (kbp), GC content (black), GC skew (purple/green). The remaining rings show BLASTn comparison of 10 complete genomes following as labelled. X. euvesicatoria (NZ_CP018467) was used as reference genome to compare the other genomes and generate the ring image using BRIGS. (B) Dendrogram shows Average Nucleotide Identity (ANI) among all genomes included in ring image. Xanthomonas species grouped in one cluster suggest that despite the similar symptoms caused by BS pathogens (X. euvesicatoria, X. vesicatoria, X. gardneri and X. perforans), they were clustered in two subgroups. X. euvesicatoria and X. perforans are the most closed related pathogens inside the BS cluster likewise X. vesicatoria and X. gardneri. No plasmid sequences were included in the analyses.
Mauve-based progressive whole genome alignments enabled the gene selection. A unique gene, ATP-dependent DNA helicase (recG) was identified and used to design LAMP primers for X. euvesicatoria. Location of the gene is indicated in Fig. 1A. Designed primers showed 100% query coverage and 100% similarity with only X. euvesicatoria sequence in NCBI GenBank databases.
Confirmation and phylogenetics of tested strains
Both sense and anti-sense strands of all the X. euvesicatoria strains used in LAMP assay validation along with other xanthomonads except X. axonopodis pv. allii and X. albilineans (Table 1) were sequenced using forward hrcN-F and reverse hrcN-R primers to confirm the identity of each strain. Manually corrected and proof-read consensus sequences of ~756 bp were aligned against the NCBI GenBank nucleotide database; obtained outcomes confirmed their identity as mentioned in Table 1. Sequences of X. euvesicatoria and other species of bacteria showed 99–100% homology to the corresponding bacterial species. Sequences of two strains A3477 and A3479 from culture collection showed 100% sequence similarity to X. axonopodis pv. glycines but they were received in the PBC as X. campestris pv. vesicatoria. Later, both strains were used in the exclusivity panel (Table 1). The phylogenetic tree showed a tight cluster of X. euvesicatoria strains in contrast to X. vesicatoria (Fig. 2). Similarly, no difference (100% homology) in pairwise identity of X. euvesicatoria strains was observed when color-coded pairwise identity matrix was generated using hrcN gene sequences (Fig. 3). All sequences were submitted to NCBI GenBank database and accession numbers are provided in Table 1.

Phylogenetic analyses of Xanthomonas euvesicatoria isolates using type III secretion system cluster gene hrcN. All isolates of X. euvesicatoria were grouped together and showed no genetic differences despite their different geographical origins.

Color-coded matrix showing pairwise identity of Xanthomonas euvesicatoria strains with strains of other species. Xv - X. vesicatoria; Xp - X. perforans, Xg - X. gardneri, X. campestris pv. campestris - Xag - X. axonopodis pv. glycines, Xad - X. axonopodis pv. diffenbachiae and Xcc - X. citri subsp. citri.
LAMP assays specificity validation
Specificity of the developed LAMP assay was confirmed using 39 strains of X. euvesicatoria, 17 strains of X. vesicatoria, X. gardneri and X. perforans, and 24 strains of other bacterial species. Additionally, genomic DNA extracted from six plants inoculated with X. vesicatoria strains were included into the exclusivity panel as well. LAMP protocols were validated for both BioRangerTM and colorimetric based detection. SYBR Green dye was added after reaction completion; positive amplification turned the dye color from orange to green and was visualized with the naked eye; florescence was observed under UV. Positive amplifications were indicated by the sigmoid shaped curve on the BioRangerTM. In the inclusivity test, all 39 X. euvesicatoria strains were specifically detected by LAMP primers. No cross reactivity was observed when X. euvesicatoria LAMP assay was tested against all 41 strains in the exclusivity panel and against the symptomatic tomato plants DNA inoculated with X. vesicatoria. No sigmoid curve, no change in color and no fluorescence under UV were observed with non-target pathogens DNA and non-template control (Table 1).

Sensitivity of Xanthomonas euvesicatoria specific loop-mediated isothermal amplification assay. (A–D) Detection of serially diluted (1 ng to 1 fg) X. euvesicatoria genomic DNA (1–7). (E–H) Detection of serially diluted (1 ng to 1 fg) X. euvesicatoria genomic DNA (1–7) spiked with 1 µl of host genomic DNA. Serially diluted DNA from 1 ng to 1 fg is represent by number 1–7. (A,E) Sensitivity assays performed using BioRangerTM, positive results are represented with a sigmoid curve; (B,F) visual observation of LAMP sensitivity results after addition of SYBR Green dye in amplified LAMP products, green color represent the positive amplification of X. euvesicatoria while orange color depict no amplification; (C,G) SYBR Green dye results under UV, positive detection resulted on fluorescence display; (D,H) agarose gel electrophoresis of LAMP product on 1.5% agarose gel. L = DNA marker; Lane 8 = non-template control.
LAMP assay sensitivity
The limit of detection and efficiency of the developed LAMP assay for X. euvesicatoria was performed using 10-fold serially diluted genomic DNA; assay detected down to 100 fg (equivalent to about 18 genome copies based on genome size and GC content, Supplemental Table 1) of genomic DNA (Fig. 4A–D). However, addition of 1 µl of host genomic DNA derived from healthy tomato plant leaves to each 10-fold serially diluted genomic DNA of X. euvesicatoria reduced the sensitivity to 1,000 fg (Fig. 4E–H). The lowest detectable amount of genomic DNA i.e. 100 fg was detected in less than 15 minutes using a portable, battery operated BioRangerTM instrument. Positive amplifications were cross confirmed using SYBR Green dye and agarose gel electrophoresis (Fig. 4B–H). A NTC was included in each run – no false negative nor false positive results were detected.
Detection of X. euvesicatoria in artificially infected plant tissue
Six-week-old healthy looking tomato plants were inoculated with six strains of X. euvesicatoria and six strains X. vesicatoria. Leaf samples were collected from symptomatic plants with typical bacterial spot symptoms that included necrotic lesions surrounded by a yellow halo on leaves and water soaked lesions on stems 10 days after inoculation. DNA was extracted from the infected and control plants and used for the X. euvesicatoria-specific LAMP assay. All six X. euvesicatoria infected tomato plant samples were positive for X. euvesicatoria (Fig. 5). The results were in agreement with results following addition of the SYBR Green dye. No positive amplification was observed when LAMP primers were tested with either healthy tomato plants or leaf samples infected with X. vesicatoria.

Detection of Xanthomonas euvesicatoria from infected samples. X. euvesicatoria was detected from infected tomato plant tissues. (A) Visual observation of LAMP results after addition of SYBR Green dye in amplified LAMP product; (B) LAMP results after addition of SYBR Green dye under UV. Tube 1 is a positive control (A6260), tube 2–7 are infected plant samples with A1781, A1706, A3478, A1788, A1718 and A1786, respectively, tube 8 is healthy plant tissue and tube 9 is non-template control (NTC; water).
Multi-operator validation tests
Multi-operator validation tests were performed by two different operators with four blind samples to confirm robustness of the developed assays. All four DNA samples were tested with LAMP assay to specifically detect X. euvesicatoria. All results obtained from both operators were in 100% agreement with the previously obtained results. No false positives or false negatives were detected during the validation test.
Discussion
In this study, we developed and validated a BioRangerTM and colorimetric based LAMP protocol for specific, sensitive, reliable and robust detection and differentiation of X. euvesicatoria, a causal agent of bacterial spot disease affecting both tomato and pepper. Nucleic acid, biochemical, and symptom based diagnostic methods for all four BS causing xanthomonads are available9,13,14,29. However, these protocols are time consuming, require skilled personnel to perform the tests, and are not point-of-care assays.
Recent advances in next generation sequencing methods have provided the framework to search for signature gene sequences to design highly specific, reliable and robust field-deployable assays30. The comparative genome analyses of ten genomes of closely related pathogens retrieved from publicly available database facilitated the identification of unique gene sequences present in X. euvesicatoria (Fig. 1). The use of MAUVE to analyze the large-scale evolutionary events among the Xanthomonas species led to the identification of a gene, recG, unique to X. euvesicatoria. Thermodynamically competent primers31 were designed to target the recG gene and validated in silico against the NCBI GenBank nucleotide database for specificity, robustness and higher accuracy (Table 2). None of the six primers showed 100% homology with any existing sequence in the database except for X. euvesicatoria (Table 2). The diagnostic assays developed using unique genes/regions of target pathogen delivered higher specificity and reliability with no possibility of cross-reaction with any other closely/distinct species compared to the assays developed using highly conserved genes present among bacterial species, like 16 s ribosomal RNA30.
The developed LAMP assay for X. euvesicatoria has been validated for specificity against X. perforans, X. vesicatoria and X. gardneri since these Xanthomonas species produce similar disease symptoms and are associated with similar hosts3,5. The X. euvesicatoria specific LAMP assay only detected X. euvesicatoria and differentiated it from closely related species, X. perforans, and the more distantly related species, X. vesicatoria and X. gardneri and all the other species tested in the exclusivity panel (Table 1). The assay was tested against strains in the inclusivity panel collected from different geographical regions to confirm their broad range detection capabilities, which makes the LAMP assay more reliable and universal so that it can be used for a wide range of applications in different parts of the world.
Compared to conventional nucleic acid-based methods, LAMP is rapid and avoids the need of sophisticated laboratory equipment like PCR and qPCR machines25. With the forward and backward loop primers, results can be obtained even in less than 20 minutes. There are numerous chemistries and several instruments used for LAMP detection from colorimetric, lateral flow device, portable battery-operated instruments to qPCR25,32. We used a field deployable battery operated small (D = 8 cm × W = 14 cm × H = 7 cm) BiorangerTM instrument for the real-time detection of reaction amplification that makes the assay easy to use for field applications. The reliability of the developed assay was confirmed by adding SYBR Green dye to the LAMP product after amplification. Despite several DNA-based detection single or multiplex PCR or quantitative Real-Time assays reported8,9,14, the current LAMP assay has enormous applications in point-of-care diagnostics without the need of bacteria isolation or sophisticated equipment. Furthermore, the reported method is highly specific and reliable to detect X. euvesicatoria from both purified bacterial DNA and infected plant material demonstrating high efficiency of the developed LAMP assay.
The sensitivity of the developed LAMP assay was evaluated to confirm the limit of detection with and without the presence of host DNA. The X. euvesicatoria LAMP assay detected pathogen genomic DNA down to 100 fg. The sensitivity of LAMP varies from pathogen to pathogen, possibly the result of bacterial functional characteristics such as extracellular polysaccharide-producing and non-producing bacteria. Polysaccharides have the capacity to inhibit DNA amplification33. Lang et al.34 reported LAMP sensitivity of 10 pg for X. oryzae pv. oryzae while it was 1 fg for X. oryzae pv. oryzicola; they interpreted that the variation in sensitivity was perhaps due to the differences in primer annealing efficiency. Similarly, reduced ability to detect target DNA in spiked assays possibly resulting from inhibitors present in host DNA35. Given that reproducibility is an essential and critical property of a diagnostic assay36, multi-operators performed the X. euvesicatoria specific LAMP assays and obtained consistent results. Hence, the developed LAMP protocol can be used in different labs without the need of standardization.
The developed LAMP assay for X. euvesicatoria detected the target pathogen in infected plant tissues with no false positive or false negative outcomes and thus can be used at point-of-care for the direct detection of the pathogen. This eliminates the necessity of culturing the pathogen, which is often a time-consuming step. The developed LAMP assay for X. euvesicatoria has the potential to be used for routine diagnostics, surveillance, biosecurity disease management and epidemiological studies. This can also be an easy-to-use tool for discovering reservoir hosts of X. euvesicatoria.
Materials and Methods
Source of isolates, plant inoculation and DNA isolation
Thirty-nine strains of X. euvesicatoria collected from many different geographical regions of the world were used in an inclusivity panel to validate the specificity of the developed LAMP assay (Table 1). Strains previously stored at −80 °C in the Pacific Bacterial Collection (University of Hawaii at Manoa) were grown on a peptone-dextrose medium containing tetrazolium chloride (5 g peptone, 2.5 g dextrose, 8.5 g agar 0.5 ml 1% TZC in 500 ml of distilled water) and a single colony was picked and grown out to preclude contamination. In addition, strains from different genera and species including X. vesicatoria, X. perforans, X. gardneri, X. citri subsp. citri, X. axonopodis pv. glycines, X. axonopodis pv. dieffenbachiae, X. axonopodis pv. allii, X. albilineans, X. citri subsp. citri, X. campestris pv. campestris, Dickeya zeae, D. diffenbachiae, D. chrysanthemi, D. solani, D. dadantii, Ralstonia solanacaerum, Clavibacter michiganensis subsp. michiganensis, C. michiganensis subsp. nebraskanensis, Pectobacterium carotovorum subsp. carotovorum, P. atrosepticum, Rathayibacter rathayi, Curtobacterium flaccumfaciens pv. poinsettiae, Pantoea ananatis, Pseudomonas syringae pv. syringae and Agrobacterium tumefaciens were included in an exclusivity panel (Table 1). All X. euvesicatoria, X. perforans, X. vesicatoria and X. gardneri strains were isolated from either tomato or pepper (Table 1).
DNA was isolated from infected and healthy plant material, and pure bacterial cultures using Wizard Genomic DNA Purification Kit (Promega, Madison, WI) and Ultra Clean Microbial DNA Isolation Kit (Mo Bio., Carlsbad, CA) following manufacturer’s instruction. DNA was quantified using NanoDropTM 2000/c Spectrophotometers (Thermo Fisher Scientific, Waltham, MA).
Six strains of X. euvesicatoria A1706, A1718, A1781, A1786, A1788 and A3478 and six of X. vesicatoria A1696, A1703, A1705, A3616, A1887 and A3618 were used to inoculate 3-weeks old tomato seedlings using foliar spray inoculation method described by Giovanardi, et al.37. X. euvesicatoria and X. vesicatoria strains were grown in YDC for 36 h at 26 ± 2 °C and water suspension was prepared for inoculation. Each inoculated plant was covered in polyethylene (PE) bag for 30 h in order to maintain the humidity and to facilitate the pathogen infection. Three weeks after inoculation, the leaves from symptomatic plants were collected. Forty milligram of leaf tissue was taken and cut in to small pieces using a sterile razor blade and placed in a 2 ml tube. After adding 600 µl of Nuclei Lysis Solution, 2 ml crew tubes were vigorously mixed using a Mini-Bead Beater 16 Center Bolt (Biospec products, Bartlesville, OK) at a maximum speed for one minute and genomic DNA extraction was performed using the Wizard Genomic DNA Purification kit following the manufacturer’s instruction. Genomic DNA isolated from healthy leaf tissue served as negative control.
Sequencing, phylogenetics and identity confirmation
Four genomes of X. euvesicatoria (NZ_CP018467), X. vesicatoria (NZ_CP018725), X. perforans (NZ_CP019725) and X. gardneri (NZ_CP018731) were retrieved from NCBI GenBank Genome Database (Supplement Table 1) and aligned with progressive Mauve38; Geneious (version 10.1.3) was used to evaluate the aligned genome regions to identify a gene that can effectively discriminate among X. euvesicatoria, X. vesicatoria, X. perforans and X. gardneri by sequencing (Supplemental Fig. 1). A gene, hrcN, from type III secretion system (T3SS) was selected for accurate identification9. A primer set hrcN-F (5′-TCGGCACCATGCTCAAGGT-3′) and hrcN-R (5′-GTGTAGAACGCGGTGATCGA-3′) was designed using Primer3 following the parameters described by Arif and Ochoa-Corona31,39. PCR conditions were as follows: Initial denaturation at 94 °C for 5 min followed by 35 cycles of denaturation at 94 °C for 20 sec, annealing 60 °C for 30 sec, extension 72 °C for 1 min and final extension at 72 °C for 3 min. PCR products were cleaned by adding 2 µl ExoSAP-IT™ (Affymetrix Inc, Santa Clara, CA) in 5 µl of PCR product and incubated at 37 °C for 15 min followed by 80 °C for 15 min. Both sense and anti-sense strands were sequenced using hrcN-F and hrcN-R primers. Sanger sequencing was performed at GENEWIZ facility (Genewiz, La Jolla, CA). Obtained sense and anti-sense strands of each isolate were aligned and manually edited to rectify any sequencing hiccups. Manually edited sequences were used to confirm the identity of each strain by comparing the sequences against the NCBI GenBank nucleotide and genome databases using NCBI BLASTn tool. Sequences were aligned, and a tree was generated with NJ tree building method using the Tree Builder module of Geneious 10.2.3. Bootstrap resampling method with 1000 replicates was used to generate the consensus tree40. Color-coded matrix showing pairwise identity was generated using Sequence Demarcation Tool v1.2.
Target selection and LAMP primer design
Genomes of X. euvesicatoria (NZ_CP018467), X. vesicatoria (NZ_CP018725), X. gardneri (NZ_CP018731) and X. perforans (NZ_CP019725), X. campestris pv. campestris (NZ_CP012145), D. solani (NZ_CP015137), X. axonopodis pv. glycines (NZ_CP017188), X. axonopodis pv. dieffenbachiae (NZ_CP014347), P. carotovorum subsp. carotovorum (NC_018525) and R. solanacearum (NC 003295) were retrieved from the NCBI GenBank genome database (Supplemental Table 1). Whole genomes were aligned with progressiveMauve. Genomes were analyzed using Geneious (10.2.3) to discover exclusive and unique gene regions in X. euvesicatoria; ATP-dependent DNA helicase recG was selected to design specific LAMP primers for X. euvesicatoria. Sense and anti-sense primer design corresponding to inner (FIP and BIP) and outer (F3 and B3) primers was carried out using PrimerExplorer V5 (https://primerexplorer.jp/e/); internal loop primers (LF and LB) were designed manually as recommended (Table 2). Specificity of each primer was confirmed in silico by screening the corresponding sequences using BLASTn tool against the NCBI nucleotide and genome databases. Locations of target genome region in X. euvesicatoria was pinpointed using BLAST Ring Image Generator (BRIG)41; ncbi-blast 2.6.0+ database was used to compare and generate BRIG image.
LAMP reaction and analyses
The six primers consisted of one pair each of outer primers (F3 and B3), inner primers (FIP and BIP) and internal loop primers (LB and LF) targeting recG gene of X. euvesicatoria were used in LAMP reaction (Table 2). LAMP reactions were carried out in a total of 25 µl reaction volume containing 2 \(\mu \)l primer mix containing 0.2 µM of each XeRec-F3/B3, 0.4 µM of each XeRec-LF/LB and 1.6 µM of each XeRec-FIP/BIP per reaction 15 \(\mu \)l Optigene® Master Mix (Optigene, West Sussex, UK), 1 \(\mu \)l template DNA and 7 \(\mu \)l water. The reaction mixture was incubated and amplified using BioRangerTM (Diagenetix Inc, Honolulu, HI), a battery-operated small unit at 65 °C for 20 min followed by melt curve analysis at 98–80 °C with an increment of 0.05 °C/sec. The obtained results were cross validated by adding 3 \(\mu \)l SYBR Green I (Molecular Probes Inc.) in each amplified reaction. Results with SYBR Green dye were visualized with naked eyes and also under UV light (FOTO/UV® 26 transilluminator, Fotodyne Inc., WI).
Sensitivity assay
Sensitivity of LAMP assay was assessed using 10-fold serially diluted purified genomic DNA of X. euvesicatoria from 1 ng to 1 fg. In addition, a spiked sensitivity assay was performed by adding 1 µl of host (tomato) genomic DNA to each serially diluted X. euvesicatoria genomic DNA samples. Non-template control (NTC; water) was included in each LAMP run.
Multi-operator validation test
Multi-operator tests were performed by two independent operators to assess the robustness of the developed X. euvesicatoria LAMP assay. Each operator performed a blind test with four samples. LAMP assay conditions and components were followed as described above.
Data Availability
All sequencing data is available in NCBI GenBank database.
References
Jones, J. B., Lacy, G. H., Bouzar, H., Stall, R. E. & Schaad, N. W. Reclassification of the xanthomonads associated with bacterial spot disease of tomato and pepper. Syst. Appl. Microbiol. 27, 755–762 (2004).
Barak, J. D. et al. Whole-Genome Sequences of Xanthomonas euvesicatoria Strains clarify taxonomy and reveal a stepwise erosion of Type 3 effectors. Front. Plant Sci. 7 (2016).
Louws, F. J. et al. Field control of bacterial spot and bacterial speck of tomato using a plant activator. Plant Dis. 85, 481–488 (2001).
Schwartz, A. R. et al. Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front. Microbiol. 6 (2015).
Ritchie, D. F. Bacterial spot of pepper and tomato. The Plant Health Instructor, https://doi.org/10.1094/PHI-I-2000-1027-01 (2000).
Potnis, N. et al. Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics 12 (2011).
Kositcharoenkul, N., Chatchawankanphanich, O., Bhunchoth, A. & Kositratana, W. Detection of Xanthomonas citri subsp. citri from field samples using single-tube nested PCR. Plant Pathol. 60, 436–442 (2011).
Moretti, C., Amatulli, M. T. & Buonaurio, R. PCR-based assay for the detection of Xanthomonas euvesicatoria causing pepper and tomato bacterial spot. Lett. Appl. Microbiol. 49, 466–471 (2009).
Strayer, A. L. et al. A Multiplex Real-Time PCR assay differentiates four Xanthomonas species associated with bacterial spot of tomato. Plant Dis. 100, 1660–1668 (2016).
Cho, M. S. et al. Sensitive and Specific Detection of Xanthomonas oryzae pv. oryzae by Real- Time Bio-PCR using pathovar-specific primers based on an rhs family gene. Am. Phytopathol. Soc. 95, 589–594 (2011).
Mondal, K. K. et al. The reliable and rapid polymerase chain reaction (PCR) diagnosis for Xanthomonas axonopodis pv. punicae in pomegranate. African J. Microbiol. Res. 6, 5950–5956 (2012).
Adriko, J. et al. Multiplex PCR for specific and robust detection of Xanthomonas campestris pv. musacearum in pure culture and infected plant material. Plant Pathol. 61, 489–497 (2012).
Koenraadt, H. et al. Development of specific primers for the molecular detection of bacterial spot of pepper and tomato. Acta Hortic. 808, 99–102 (2009).
Araújo, E. R., Costa, J. R., Ferreira, M. A. S. V. & Quezado-Duval, A. M. Simultaneous detection and identification of the Xanthomonas species complex associated with tomato bacterial spot using species-specific primers and multiplex PCR. J. Appl. Microbiol. 113, 1479–1490 (2012).
Albuquerque, P. et al. Evolutionary and experimental assessment of novel markers for detection of Xanthomonas euvesicatoria in plant samples. PLoS One 7, e37836 (2012).
Duan, Y. et al. Development and application of loop-mediated isothermal amplification for detection of the F167Y mutation of carbendazim-resistant isolates in Fusarium graminearum. Sci. Rep. 4, 1–8 (2014).
Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. DNA detection using recombination proteins. PLoS Biol. 4, e204 (2006).
Little, M. C. et al. Strand displacement amplification and homogeneous real-time detection incorporated in a second-generation DNA probe system, BDProbeTecET. Clin Chem 45, 777–784 (1999).
Vincent, M., Xu, Y. & Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 5, 795–800 (2004).
Van Ness, J., Van Ness, L. K. & Galas, D. J. Isothermal reactions for the amplification of oligonucleotides. Proc Natl Acad Sci USA 100, 4504–4509 (2013).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, E63 (2000).
Dean, F. B., Nelson, J. R., Giesler, T. L. & Lasken, R. S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 11 (2001).
Zhang, Y. & Tanner, N. A. Isothermal amplification of long, discrete DNA fragments acilitated by Single-Stranded Binding Protein. Sci. Rep. 7, 1–9 (2017).
Yasuhara-Bell, J., Marrero, G., De Silva, A. & Alvarez, A. M. Specific detection of Pectobacterium carotovorum by loop-mediated isothermal amplification. Mol. Plant Pathol. 17, 1499–1505 (2016).
Yasuhara-Bell, J. H., Marrero, G., Arif, M., de Silva, A. & Alvarez, A. Development of a Loop-Mediated Isothermal Amplification (LAMP) assay for the detection of Dickeya spp. Phytopathology 107, PHYTO–04–17–0160–R (2017).
Nagamine, K., Hase, T. & Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probe 16, 223–229 (2002).
Kubota, R., Vine, B. G., Alvarez, A. M. & Jenkins, D. M. Detection of Ralstonia solanacearum by loop-mediated isothermal amplification. Phytopathology 98, 1045–1051 (2008).
Wen, Y.-M. & Wang, Y.-X. Biological features of hepatitis B virus isolates from patients based on full-length genomic analysis. Rev. Med. Virol. 19, 57–64 (2009).
Stall, R. E., Jones, J. B. & Minsavage, G. V. Durability of resistance in tomato and pepper to xanthomonads causing bacterial spot. Annu. Rev. Phytopathol. 47, 265–284 (2009).
Ouyang, P., Arif, M., Fletcher, J., Melcher, U. & Ochoa Corona, F. M.Enhanced reliability and accuracy for field deployable bioforensic detection and discrimination of Xylella fastidiosa subsp. pauca, causal agent of citrus variegated chlorosis using Razor Ex technology and Taqman quantitative PCR. Plos One 8, e81647 (2013).
Arif, M. & Ochoa-Corona, F. M. Comparative assessment of 5′ A/T-rich overhang sequences with optimal and sub-optimal primers to increase PCR yields and sensitivity. Mol. Biotechnol. 55, 17–26 (2013).
Ash, G. J. et al. Development of a genomics-based LAMP (loop-mediated isothermal amplification) assay for detection of Pseudomonas fuscovaginae from rice. Plant Dis. 98, 909–915 (2014).
Schrader, C., Schielke, A., Ellerbroek, L. & Johne, R. PCR inhibitors - occurrence, properties and removal. J. Appl. Microbiol. 113, 1014–1026 (2012).
Lang, J. M. et al. Sensitive detection of Xanthomonas oryzae pathovars oryzae and oryzicola by loop-mediated isothermal amplification. Appl. Environ. Microbiol. 80, 4519–4530 (2014).
Dobhal, S., Olson, J. D., Arif, M., Garcia Suarez, J. A. & Ochoa-Corona, F. M. A simplified strategy for sensitive detection of Rose rosette virus compatible with three RT-PCR chemistries. J. Virol. Methods 232, 47–56 (2016).
Arif, M., Fletcher, J., Marek, S. M., Melcher, U. & Ochoa-Corona, F. M. Development of a rapid, sensitive, and field-deployable Razor Ex biodetection system and quantitative PCR assay for detection of Phymatotrichopsis omnivora using multiple gene targets. Appl. Environ. Microbiol. 79, 2312–2320 (2013).
Giovanardi, D., Biondi, E., Ignjatov, M., Jevtić, R. & Stefani, E. Impact of bacterial spot outbreaks on the phytosanitary quality of tomato and pepper seeds. Plant Pathology 67, 1168–1176 (2018).
Darling, A. E., Mau, B. & Perna, N. T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, (2010).
Rozen, S., & Skaletsky, H. J. Primer3 on the www for general users and for biologist programmers. In Krawetz, S. & Misener, S. (Eds), Bioinformatics methods and protocols: Methods in Molecular Biology (pp. 365–386). Totowa, NJ: Humana Press. (2000).
Felsenstein, J. Confidence Limits on Phylogenies: An approach using the bootstrap Author (s): Joseph Felsenstein Published by: Society for the Study of Evolution Stable. Evolution (N. Y). 39, 783–791 (1985).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genomics 12 (2011).
Acknowledgements
This work was supported by the USDA National Institute of Food and Agriculture, Hatch project 9038H, managed by the College of Tropical Agriculture and Human Resources and the National Science Foundation (NSF-CSBR Grant No. DBI-1561663).
Author information
Authors and Affiliations
Contributions
M.A. conceived and designed the study. A.L., U.D., G.B. and L.F. performed the experiments and wrote the manuscript. A.A. collected and initially purified the strains used in this study. M.A. and A.A. revised the manuscript and provided ideas and support for the final submission. All authors reviewed and aproved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Larrea-Sarmiento, A., Dhakal, U., Boluk, G. et al. Development of a genome-informed loop-mediated isothermal amplification assay for rapid and specific detection of Xanthomonas euvesicatoria. Sci Rep 8, 14298 (2018). https://doi.org/10.1038/s41598-018-32295-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32295-4
- Springer Nature Limited
Keywords
This article is cited by
-
Specific colorimetric LAMP assay for the detection of maize bushy stunt phytoplasma in corn through comparative genomics
Tropical Plant Pathology (2024)
-
Mayaro virus detection by integrating sample preparation with isothermal amplification in portable devices
Analytical and Bioanalytical Chemistry (2023)
-
Novel plant disease detection techniques-a brief review
Molecular Biology Reports (2023)
-
Development of a reverse transcription-loop mediated isothermal amplification assay for detection of wisteria vein mosaic virus
European Journal of Plant Pathology (2022)
-
Multiplex recombinase polymerase amplification assay developed using unique genomic regions for rapid on-site detection of genus Clavibacter and C. nebraskensis
Scientific Reports (2021)