
Abstract
Lupus nephritis (LN) is a common and serious clinical manifestation of systemic lupus erythematosus. However, the pathogenesis of LN is not fully understood. The currently available treatments do not cure the disease and appear to have a variety of side effects in the long term. The purpose of this study was to search for key molecules involved in the LN immune response through bioinformatics techniques to provide a reference for LN-specific targeted therapy. The GSE112943 dataset was downloaded from the Gene Expression Omnibus database, and 20 of the samples were selected for analysis. In total, 2330 differentially expressed genes were screened. These genes were intersected with a list of immune genes obtained from the IMMPORT immune database to obtain 128 differentially expressed immune-related genes. Enrichment analysis showed that most of these genes were enriched in the interferon signalling pathway. Gene set enrichment analysis revealed that the sample was significantly enriched for expression of the interferon signalling pathway. Further analysis of the core gene cluster showed that nine genes, GBP2, VCAM1, ADAR, IFITM1, BST2, MX2, IRF5, OAS1 and TRIM22, were involved in the interferon signalling pathway. According to our analysis, the guanylate binding protein 2 (GBP2), interferon regulatory factor 5 and 2′-5′-oligoadenylate synthetase 1 (OAS1) genes are involved in three interferon signalling pathways. At present, we do not know whether GBP2 is associated with LN. Therefore, this study focused on the relationship between GBP2 and LN pathogenesis. We speculate that GBP2 may play a role in the pathogenesis of LN as a member of the interferon signalling pathway. Further immunohistochemical results showed that the expression of GBP2 was increased in the renal tissues of LN patients compared with the control group, confirming this conjecture. In conclusion, GBP2 is a member of the interferon signalling pathway that may have implications for the pathogenesis of LN and serves as a potential biomarker for LN.
Similar content being viewed by others


Introduction
SLE is a common chronic, multisystem, autoimmune disease of unknown aetiology, the causes of which may include environmental and stochastic factors and genetic susceptibility [1, 2]. LN is a common and severe clinical manifestation of SLE [3,4,5] and a major risk factor for mortality [6]. Approximately 31–48% of patients with SLE develop LN. Furthermore, 7–31% of SLE patients are diagnosed with LN at the time of SLE diagnosis [7,8,9]; of these patients, 10% progress to end-stage renal disease (ESRD)[4, 5]. Patients with LN have a higher risk of death than the general population, and the risk of death is further increased if LN progresses to end-stage renal disease [10].
Previous studies on the pathogenesis of LN have focused on the adaptive immune system. It is usually assumed that lymphocyte abnormalities are the main cause of autoimmunity due to the recognition and processing of autoantigens by the immune system. These autoantigens activate the IFN-I signalling system and the resulting immune response in an organism by a series of antibodies that recognize autoantigens [11, 12]. There is evidence that the clinical manifestations of SLE are caused by biological responses triggered by the overproduction of IFN-I [13]. A variety of immune cells are affected by IFN-I, as IFN-I regulates intermediate signalling substances required for multiple cytokine responses [14]. IFN-stimulated genes (ISGs) are induced by IFN-I expression, and increased expression of IFN-inducible protein (IIP) in immune cells and its altered function can promote SLE disease progression [15]. Glucocorticoids were previously reported to improve patient survival, but treatment outcomes remain unsatisfactory [16, 17]. Although immunosuppressive therapy may alleviate this disease, recurrent episodes of the disease continue to damage the kidneys and eventually progress to ESRD [18]. In this context, there is an extremely urgent need to explore new therapies that are more effective, more targeted, and safer. Therefore, further exploration of the aetiology and pathogenesis of LN is necessary to find specific drugs for the effective treatment of this disease and to further improve the survival of patients.
In recent years, bioinformatics and microarray technologies have rapidly developed. Furthermore, bioinformatics has been widely used to identify disease-related genes and analyse disease pathogenesis, helping to identify important molecules associated with diseases and their mechanisms of action [19,20,21]. In this study, we used a bioinformatics approach to analyse genetic data from LN kidney tissues and normal control kidney tissues to screen for differentially expressed genes (DEGs) in LN. The aim of this study was to identify biomarkers associated with LN disease and to explore their pathways of action.
Results
Screening of 128 differentially expressed immune-related genes
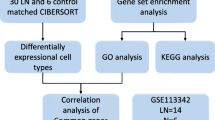
Figure 1 shows the specific databases used in this study and the complete workflow. Based on the sample information, 2330 differentially expressed genes were extracted from the LN samples, of which 2053 genes were upregulated, and 277 genes were downregulated. The screening criteria for differentially expressed genes were as follows: the fold change between the LN group and the control group was at least eightfold, and the corrected P value was < 0.05. To better display these differentially expressed genes, a heatmap and volcano map were drawn using R language. Heatmaps were created using the pheatmap package (Fig. 2a), and volcano plots were created using the ggplot2 package (Fig. 2b). These differentially expressed genes intersected with the list of immune genes obtained from the IMMPORT immune database to obtain 128 differentially expressed immune-related genes (Fig. 2c). These genes included 111 upregulated genes and 17 downregulated genes (Table 1).

Study workflow diagram. DEGs: differentially expressed genes. GSEA: gene set enrichment analysis

a Heat map of differentially expressed genes between LN and HC samples. Red rectangles indicate high expression and purple rectangles indicate low expression. hc: healthy control. b Volcano plot showing differentially expressed genes in LN and HC samples, blue dots represent genes significantly down-regulated in the samples and red dotes represent genes significantly up-regulated. hc: healthy control group. c Venn diagram showing 128 differentially immune-related genes obtained by intersecting differentially expressed genes and immune genes. DEGs: differentially expressed genes. IGs: immune genes
Differentially expressed immune-related genes are significantly enriched in the interferon signalling pathway
To identify the signalling pathways with which these 128 differentially expressed immune-related genes are involved, we further analysed the pathways enriched in these genes. The results of the DAVID software enrichment analysis showed that these genes are mainly involved in the immune response, signal transduction and microbial infection pathways (Fig. 3a). The results of the FUNRICH enrichment analysis showed that these genes were mainly enriched in the interferon signalling pathway, cytokines and the cell membrane signalling pathway (Fig. 3b).

a Enrichment analysis of differential immune-related genes using DAVID software, with the top 8 biological pathways selected based on enrichment scores, shown using bubble plots. P < 0.05 is statistically significant. b Enrichment analysis of differential immune-associated genes using Funrich software, with the top 6 biological pathways selected based on P-value and gene percentage, shown using bar graphs. P < 0.05 was statistically significant. c Validation of the results of enrichment analysis of the differential immune-related genes using metascape software, a total of 20 pathways were enriched, shown using bar graphs. p < 0.05 was statistically significant. d Hallmarks gene set base used to analyse the entire gene expression values of LN and HC smaples. Significant enrichment in the interferon alpha pathway is shown, p < 0.05. e Hallmarks gene set database used to analyze the entire gene expression value of LN and HC samples. Shows significant enrichment in the interferon gamma pathway. p < 0.05
Finally, the enrichment results were validated using METASCAPE software. The results revealed 20 pathways, including the cytokine signalling pathway, activation of immune cells and interferon signalling pathway in the immune system (Fig. 3c). The enrichment analysis results showed that these genes were significantly enriched in the interferon signalling pathway.
The interferon signalling pathway is significantly enriched
To understand the overall gene expression, we performed GSEA for all gene expression information of the LN group and the control group using the CLUSTERPROFILER [22] software package based on the hallmark and KEGG gene set databases [23]. The default value for significantly enriched gene sets was set to a corrected P value < 0.05. GSEA revealed that the sample expression information was significantly enriched in the interferon α/γ response (Fig. 3d-e).
PPI networks reveal four core clusters of differentially expressed immune-associated genes.
We further searched for core gene clusters among these 128 differentially expressed immune-related genes and analysed these genes using STRING to obtain protein interaction network maps. The network maps were visualized by CYTOSCAPE. The network map generated 116 nodes and 664 connecting lines (Fig. 4a). Data were processed with the MCODE (degree cut-off = 2, node score cut-off = 0.2, k-core = 2, maximum depth = 100) plugin to select the gene clusters (Table 2). Among these clusters, 4 gene clusters were obtained (Fig. 4b-e).

Processing the protein interaction network with Cytoscape v3.8.2. a The difference clusters of the MCODE analysis are indicated by different colors. b Gene cluster 1 (score: 6.667, 19 nodes, 120 edges). c Gene cluster 2 (score: 6.364, 23 nodes, 140 edges). d Gene cluster 3 (score: 5.294, 18 nodes, 90 edges). e Gene cluster 4 (score: 3, 3 nodes, 6 edges)
GBP2, IRF5 and OAS1 are involved in three interferon signalling pathways
We selected the highest scoring gene clusters for analysis to find the core genes among them. The results of GO analysis showed that these genes are mainly involved in defence responses to viruses and in the biological processes of the interferon signalling pathway (Fig. 5a). The results of STRING analysis showed that nine genes, GBP2, VCAM1, ADAR, IFITM1, BST2, MX2, IRF5, OAS1 and TRIM22, are involved in the interferon signalling pathway, three of which (GBP2, IRF5 and OAS1) are involved in three interferon signalling pathways (Fig. 5b). IRF5 is known to induce IFN expression and plays an important role in the pathogenesis of SLE [22, 23] [23, 24]. The literature also reports that OAS1 is associated with the pathogenesis of SLE [25] [26]. However, it is not clear whether GBP2 is associated with LN disease.

a GO analysis of the highest scoring gene clusters using the R language cluster Profiler package, which showed that these genes are mainly involved in biological processes such as defense responses to viruses and the interferon signalling pathway. b Reactome pathway results showing genes involved in the interferon signaling path way using STRING for the highest scoring gene clusters. The figures shows the genes involved in each pathway
GBP2 expression increases in LN in the GSE32592 dataset
To clarify the expression of GBP2 in LN, we used the GSE32592 dataset for analysis. Based on GSE32592, we first analysed the overall expression of GBP2 in kidney tissues, and the results showed that its expression was increased in LN compared with normal kidney tissues (Fig. 6a). Then, we verified the expression of GBP2 in glomeruli and tubulointerstitium, and the results also showed that the expression of GBP2 was increased in LN (Fig. 6b-c).

Expression of GBP2 in dataset GSE32592. a Expression in the kidney tissue. b expression in the glomeruli. c expression in the tubulointerstitium
GBP2 expression is significantly higher in the LN group than in the control group
To further validate the expression of GBP2 in LN, we analysed the expression of GBP2 in the LN group versus the control group using immunohistochemistry (Fig. 7a). Consistent with this prediction, the results showed that the expression of GBP2 in the LN group was significantly higher than that in the control group (Fig. 7b). The overall mean GBP2 expression was significantly different between the LN group and control groups (difference 25.565, CI 19.773–31.358, P < 0.001). The detailed clinical information of the patient was shown in Additional file 1: Table S1.

a Expression of GBP2 in LN and control. b GBP2 expression box plots for the LN and control groups, showing a statistical difference in difference in overall means (difference 25.565, CI 28.565, CI 19.773–31.358, P < 0.001)
Discussion
SLE is a chronic autoimmune disease that is characterized by multiple autoantibodies and involves both the innate and adaptive immune systems [27]. LN is a common clinical presentation of SLE [3,4,5], and its pathogenesis includes autoantibody production, abnormal activation of innate and adaptive immune responses, and immune-mediated renal injury [28]. Early studies on the pathogenesis of LN focused on the adaptive immune system. However, the molecular mechanisms underlying the pathogenesis of LN are still not fully understood, and no specific drugs have been identified to effectively treat this disease.
In recent years, the discovery of several molecules closely related to the pathogenesis of LN has greatly contributed to a new understanding of the disease and new therapeutic directions. However, the pathogenesis is still not fully understood at the molecular level, which has greatly hindered new advances in therapeutic approaches to the disease. With the development of single-cell sequencing technology and bioinformatics technology, an increasing number of genes associated with the pathogenesis of LN have been discovered [29, 30]. These genes offer the possibility to explore new targets for LN therapy.
In this study, we screened the differentially expressed genes from a dataset downloaded from the GEO database and analysed their intersection with the immune genes downloaded from the IMMPORT database. Overall, 128 differentially expressed immune-related genes were screened, including 111 upregulated genes and 17 downregulated genes. We used the DAVID, FUNRICH, and METASCAPE databases for enrichment analysis of the 128 differentially expressed immune-related genes, and we found that the interferon signalling pathway was significantly enriched. To validate the results, we analysed the overall expression information of the samples by the CLUSTERPROFILER package in R language, and the results showed that the overall expression was also significantly enriched in the interferon signalling pathway.
Persistent overexpression of interferon and its continuous stimulation of the immune system are responsible for various clinical manifestations of SLE [31]. Previous studies have shown that activation of the IFN signalling pathway is associated with LN [32] and active LN [33]. Studies have also shown that a large number of genes are regulated by interferon [31] and that LN kidney biopsies show increased expression of IFN-induced genes [34, 35].
To determine which of these 128 genes are involved in the interferon pathway, we analysed these 128 genes with MCODE and obtained the highest scoring gene cluster. We analysed this gene cluster with R software to determine the biological processes with which these genes are involved. The results showed that these genes were significantly enriched in the interferon signalling pathway. REACTOME pathway analysis showed that the genes involved in the interferon signalling pathway included nine genes: GBP2, VCAM1, ADAR, IFITM1, BST2, MX2, IRF5, OAS1 and TRIM22. Among these genes, GBP2, IRF5 and OAS1 are involved in three interferon signalling pathways. IRF5 plays a role in the pathogenesis of SLE in a variety of cells [36, 37], and OAS1 is associated with SLE pathogenesis [25]. Here, we explored the relationship between GBP2 and the pathogenesis of LN.
Guanylate binding proteins (GBPs) are IFN-inducible proteins [38]. Previous studies have shown that GBPs are mainly involved in the innate immune response to bacterial infections [39] and have an important role in protective immunity against bacterial infections [40,41,42,43,44]. Additionally, GBPs have a wide range of antiviral properties and play an important role in host resistance to viral infections [45]. GBP2 is a member of the GBP family. IFN-α/β and IFN-γ induce the production of GBP2 [46], which also plays an important role in resistance to infection by intracellular pathogens [47]. GBP2 inhibits a variety of viruses, including human immunodeficiency virus, hepatitis C, swine fever, Zika virus, measles, and influenza A [48,49,50,51]. GBP2 has been reported to induce cytoplasmic lysis and DNA release during bacterial infection and to promote the activation of melanoma infection factor 2 (AIM2) by the inflammasome [43].
AIM2 was first identified in melanoma [52] and is an interferon-inducible protein [53]. AIM2 has been shown to act as a cytoplasmic double-stranded DNA sensor. It is a component of the inflammasome that recognizes pathogen-associated or host-derived cytoplasmic double-stranded DNA. This triggers the production of interleukin 18 (IL-18) and interleukin 1-beta (IL-1β) and initiates the innate immune system [54, 55]. It has been reported in the literature that AIM2 may act as an important cytoplasmic double-stranded DNA sensor that induces the functional maturation of macrophages and serves as a potential biomarker for SLE disease [56].
Macrophages secrete a variety of cytokines; through these cytokines, they participate in the inflammatory response and regulate adaptive immunity [57]. Studies have shown that SLE patients present with abnormal cell death, including apoptosis, cell necrosis and enhanced autophagy, along with reduced clearance of dead cells [58]. It has been reported that macrophages are closely associated with poor prognosis by mediating inflammation and tissue remodelling, leading to LN tissue damage and renal macrophage infiltration [56]. These studies have demonstrated the role of macrophages in the pathogenesis of SLE. Additionally, experiments have increasingly reported a close relationship between SLE and macrophages [59, 60]. Conversely, blocking macrophage activation alleviates the progression of SLE, suggesting that apoptotic double-stranded DNA-induced macrophage activation may play an important pathogenic role in the development of SLE [56].
Conclusion
The current study suggests that GBP2 is a member of the interferon signalling pathway. We found that it may play a role in the pathogenesis of LN. Immunohistochemical results showed that the expression of GBP2 was significantly increased in LN patients compared with controls. Therefore, we suggest that GBP2, as an interferon-inducible gene, plays a role in LN disease progression, providing a new perspective on the understanding of this disease. This novel finding lays the foundation for the study of the underlying mechanisms of LN and indicates potentially promising findings for clinical application.
Materials and methods
Acquisition of sample information
We obtained human LN expression profiles from the Gene Expression Omnibus database [61], from which we selected the GSE112943 dataset based on the GPL10558 platform [62]. From this dataset, 20 kidney samples were selected, including 14 LN kidney samples and 6 control kidney samples. All biological information for the selected samples was downloaded for the next step of analysis. The sample information and data used in this paper were downloaded from public databases.
Data processing
The downloaded raw expression matrix was processed using R language to convert probe IDs into gene symbols and delete probes that could not be converted into gene symbols based on the annotation information in the platform file. When multiple probes all represented a gene symbol, the one with the highest expression was selected to represent the expression level of that gene. Differentially expressed genes were screened using the R language LIMMA package [63]. The criteria for selecting differentially expressed genes were as follows: at least eightfold change between the LN group and the control group and a corrected P value < 0.05. The obtained differential genes were intersected with the list of immune genes downloaded from the IMMPORT immune database to obtain the differentially expressed immune-related genes.
Enrichment analysis of samples
The annotation, visualization and integrated discovery database (DAVID v6.8) [64] and the functional enrichment analysis tool (FUNRICH) v3.1.3 [65] were used for pathway analysis of the differentially expressed immune-related genes. The enrichment results were validated using METASCAPE [66] software, and the differentially expressed immune-related genes were uploaded to METASCAPE for pathway analysis. The pathway analysis aimed to identify the key pathways involved in the differentially expressed immune-related genes.
Gene set enrichment analysis (GSEA) was performed using the R language CLUSTERPROFILER package [67] for all genetic information in the LN group and the control group. This analysis is based on the expression of the overall genome of the sample rather than individual genes and therefore allows for the observation of more subtle changes in expression.
Protein‒protein interaction (PPI) network analysis and gene cluster analysis
The screened differentially expressed immune-related genes were uploaded to STRING v11.5 [68] to obtain protein‒protein interaction network maps. The results of STRING analysis were imported into CYTOSCAPE v3.8.2 [69], and clustering analysis was performed using the Molecular Complex Detection (MCODE) plugin. The gene clusters with high scores were selected. We further analysed the biological processes with which the genes in that gene cluster were involved.
Enrichment analysis of the highest scoring gene clusters
The genes in the highest scoring gene clusters were subjected to gene ontology (GO) analysis using the R language CLUSTERPROFILER package to analyse the biological processes with which these genes are mainly involved. Then, these genes were further analysed with STRING to identify the genes involved in the main biological processes.
Dataset GSE32592 validates the expression of core genes in LN
We screened a potential core gene from the highest scoring gene cluster and analysed it using the GSE32592 dataset. Based on the information from GSE32592, we analysed the core genes at the overall kidney, glomerular and tubulointerstitial levels to clarify the expression of core genes in the LN.
Immunohistochemistry
To further clarify the expression of core genes in LN, they were validated by immunohistochemistry. We selected renal pathological sections from 12 patients who attended the Affiliated Hospital of Xuzhou Medical University between January 2020 and December 2021. These patients included 6 patients with active LN (5 females and 1 male, age range 21–41 years) and 6 patients with hydremic nephritis, including 5 patients with membranous nephropathy and 1 patient with minimal change disease (5 females and 1 male, age range 25–56 years). The study was approved by the Ethics Committee of the Affiliated Hospital of Xuzhou Medical University, and all patients provided written informed consent. The selected LN patients met the American College of Rheumatology (ACR) classification criteria for SLE and had biopsy-confirmed lupus nephritis. Patients also had a mean activity index (AI) > 12 based on a modified National Institutes of Health (NIH) semiquantitative score. Six patients with LN with renal lesions were used as the experimental group, and 5 patients with membranous nephropathy and 1 patient with minimal change disease were used as the control group. Immunohistochemistry was performed by KingMed Diagnostics (Nanjing Jinyu Medical Testing Center Co., Ltd, China) to analyse the expression of GBP2 (PROTEINTECH Group, Inc.) in the experimental and control groups.
Statistical analysis
The area ratios of the experimental and control groups were calculated using ImageJ software. SPSS Version 25.0 (SPSS, Armonk, NY, USA) was applied for statistical analysis of the data. Values are expressed as the means ± standard deviation, and normality was tested for the LN group and the control group using the Shapiro–Wilk test. Student’s t test was used when the variables were normally distributed in both groups, and the t’ test was used if the variance was not equal. Differences were considered statistically significant at P < 0.05.
Availability of data and materials
The data used in this study were obtained from the GEO database (GEO; http://www.ncbi.nlm.nih.gov/geo/): GSE112943.
Abbreviations
- LN:
-
Lupus nephritis
- SLE:
-
Systemic lupus erythematosus
- GEO:
-
Gene expression omnibus
- GSEA:
-
Gene set enrichment analysis
- GBP2:
-
Guanylate binding protein 2
- IRF5:
-
Interferon regulatory factor 5
- OAS1:
-
2′-5′-Oligoadenylate synthetase 1
- ESRD:
-
End-stage renal disease
- ISG:
-
IFN-inducible gene
- IIP:
-
IFN-inducible protein
- DEGs:
-
Differentially expressed genes
- ACR:
-
American College of Rheumatology
- AI:
-
Activity index
- NIH:
-
National Institutes of Health
- AIM2:
-
Activation of melanoma infection factor 2
- IL-18:
-
Interleukin 18
- IL-1β:
-
Interleukin 1-beta
References
Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–30.
Chen Z, Lan R, Ye K, Chen H, Chen C, Xu Y. Prioritization of diagnostic and prognostic biomarkers for lupus nephritis based on integrated bioinformatics analyses. Front Bioeng Biotechnol. 2021;9: 717234.
Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019;20(7):902–14.
Fava A, Buyon J, Mohan C, Zhang T, Belmont HM, Izmirly P, et al. Integrated urine proteomics and renal single-cell genomics identify an IFN-gamma response gradient in lupus nephritis. JCI Insight. 2020. https://doi.org/10.1172/jci.insight.138345.
Rao DA, Arazi A, Wofsy D, Diamond B. Design and application of single-cell RNA sequencing to study kidney immune cells in lupus nephritis. Nat Rev Nephrol. 2020;16(4):238–50.
Ding X, Ren Y, He X. IFN-I mediates lupus nephritis from the beginning to renal fibrosis. Front Immunol. 2021;12: 676082.
Mahajan A, Amelio J, Gairy K, Kaur G, Levy RA, Roth D, et al. Systemic lupus erythematosus, lupus nephritis and end-stage renal disease: a pragmatic review mapping disease severity and progression. Lupus. 2020;29(9):1011–20.
Galindo-Izquierdo M, Rodriguez-Almaraz E, Pego-Reigosa JM, Lopez-Longo FJ, Calvo-Alen J, Olive A, et al. Characterization of patients with lupus nephritis included in a large cohort from the Spanish Society of Rheumatology Registry of Patients With Systemic Lupus Erythematosus (RELESSER). Medicine (Baltimore). 2016;95(9): e2891.
Hanly JG, O’Keeffe AG, Su L, Urowitz MB, Romero-Diaz J, Gordon C, et al. The frequency and outcome of lupus nephritis: results from an international inception cohort study. Rheumatology (Oxford). 2016;55(2):252–62.
Yap DY, Tang CS, Ma MK, Lam MF, Chan TM. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol Dial Transplant. 2012;27(8):3248–54.
Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol. 2019;20(7):915–27.
Deng Y, Tsao BP. Updates in lupus genetics. Curr Rheumatol Rep. 2017;19(11):68.
Choubey D, Panchanathan R. Absent in Melanoma 2 proteins in SLE. Clin Immunol. 2017;176:42–8.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49.
Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010: 948364.
Sterner RM, Hartono SP, Grande JP. The pathogenesis of lupus nephritis. J Clin Cell Immunol. 2014. https://doi.org/10.4172/2155-9899.1000205.
Schwartz N, Goilav B, Putterman C. The pathogenesis, diagnosis and treatment of lupus nephritis. Curr Opin Rheumatol. 2014;26(5):502–9.
Chan TM. Treatment of severe lupus nephritis: the new horizon. Nat Rev Nephrol. 2015;11(1):46–61.
Yi XH, Zhang B, Fu YR, Yi ZJ. STAT1 and its related molecules as potential biomarkers in Mycobacterium tuberculosis infection. J Cell Mol Med. 2020;24(5):2866–78.
Wang H, Xie Q, Ou-Yang W, Zhang M. Integrative analyses of genes associated with idiopathic pulmonary fibrosis. J Cell Biochem. 2018. https://doi.org/10.1002/jcb.28153.
Wang H, Zhu H, Zhu W, Xu Y, Wang N, Han B, et al. Bioinformatic analysis identifies potential key genes in the pathogenesis of turner syndrome. Front Endocrinol (Lausanne). 2020;11:104.
Ban T, Kikuchi M, Sato GR, Manabe A, Tagata N, Harita K, et al. Genetic and chemical inhibition of IRF5 suppresses pre-existing mouse lupus-like disease. Nat Commun. 2021;12(1):4379.
Song S, De S, Nelson V, Chopra S, LaPan M, Kampta K, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest. 2020;130(12):6700–17.
Brune Z, Rice MR, Barnes BJ. Potential T Cell-Intrinsic regulatory roles for IRF5 via cytokine modulation in T helper subset differentiation and function. Front Immunol. 2020;11:1143.
Qing J, Song W, Tian L, Samuel SB, Li Y. Potential small molecules for therapy of lupus nephritis based on genetic effect and immune infiltration. Biomed Res Int. 2022;2022:2259164.
Fang Q, Li T, Chen P, Wu Y, Wang T, Mo L, et al. Comparative analysis on abnormal methylome of differentially expressed genes and disease pathways in the immune cells of RA and SLE. Front Immunol. 2021;12: 668007.
Wang Y, Xiao S, Xia Y, Wang H. The therapeutic strategies for SLE by targeting Anti-dsDNA antibodies. Clin Rev Allergy Immunol. 2021. https://doi.org/10.1007/s12016-021-08898-7.
Yung S, Yap DY, Chan TM. A review of advances in the understanding of lupus nephritis pathogenesis as a basis for emerging therapies. F1000Research. 2020. https://doi.org/10.12688/f1000research.22438.1.
Cao Y, Tang W, Tang W. Immune cell infiltration characteristics and related core genes in lupus nephritis: results from bioinformatic analysis. BMC Immunol. 2019;20(1):37.
Kong J, Li L, Zhimin L, Yan J, Ji D, Chen Y, et al. Potential protein biomarkers for systemic lupus erythematosus determined by bioinformatics analysis. Comput Biol Chem. 2019;83: 107135.
Ronnblom L, Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med. 2019;6(1): e000270.
Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52(5):1491–503.
Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2951–62.
Castellano G, Cafiero C, Divella C, Sallustio F, Gigante M, Pontrelli P, et al. Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and LMP7 induction in renal tubular epithelial cells. Arthritis Res Ther. 2015;17:72.
Watanabe S, Imaizumi T, Tsuruga K, Aizawa T, Ito T, Matsumiya T, et al. Glomerular expression of myxovirus resistance protein 1 in human mesangial cells: possible activation of innate immunity in the pathogenesis of lupus nephritis. Nephrology (Carlton). 2013;18(12):833–7.
Eames HL, Corbin AL, Udalova IA. Interferon regulatory factor 5 in human autoimmunity and murine models of autoimmune disease. Transl Res. 2016;167(1):167–82.
Ban T, Sato GR, Tamura T. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus. Int Immunol. 2018;30(11):529–36.
Kresse A, Konermann C, Degrandi D, Beuter-Gunia C, Wuerthner J, Pfeffer K, et al. Analyses of murine GBP homology clusters based on in silico, in vitro and in vivo studies. BMC Genomics. 2008;9:158.
Godoy P, Cadenas C, Hellwig B, Marchan R, Stewart J, Reif R, et al. Interferon-inducible guanylate binding protein (GBP2) is associated with better prognosis in breast cancer and indicates an efficient T cell response. Breast Cancer. 2014;21(4):491–9.
Huang S, Meng Q, Maminska A, MacMicking JD. Cell-autonomous immunity by IFN-induced GBPs in animals and plants. Curr Opin Immunol. 2019;60:71–80.
Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336(6080):481–5.
Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA. 2014;111(16):6046–51.
Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol. 2015;16(5):476–84.
Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509(7500):366–70.
Clough B, Finethy R, Khan RT, Fisch D, Jordan S, Patel H, et al. C57BL/6 and 129 inbred mouse strains differ in Gbp2 and Gbp2b expression in response to inflammatory stimuli in vivo. Wellcome Open Res. 2019;4:124.
Kotov DI, Mitchell JS, Pengo T, Ruedl C, Way SS, Langlois RA, et al. TCR affinity biases Th cell differentiation by regulating CD25, Eef1e1, and Gbp2. J Immunol. 2019;202(9):2535–45.
Ramsauer K, Farlik M, Zupkovitz G, Seiser C, Kroger A, Hauser H, et al. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc Natl Acad Sci USA. 2007;104(8):2849–54.
Srinivasachar Badarinarayan S, Shcherbakova I, Langer S, Koepke L, Preising A, Hotter D, et al. HIV-1 infection activates endogenous retroviral promoters regulating antiviral gene expression. Nucleic Acids Res. 2020;48(19):10890–908.
Braun E, Hotter D, Koepke L, Zech F, Gross R, Sparrer KMJ, et al. Guanylate-binding proteins 2 and 5 exert broad antiviral activity by inhibiting furin-mediated processing of viral envelope proteins. Cell Rep. 2019;27(7):2092-104 e10.
Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Sturzel CM, et al. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe. 2016;19(4):504–14.
Li LF, Yu J, Li Y, Wang J, Li S, Zhang L, et al. Guanylate-binding protein 1, an interferon-induced GTPase, exerts an antiviral activity against classical swine fever virus depending on Its GTPase activity. J Virol. 2016;90(9):4412–26.
DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA, et al. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene. 1997;15(4):453–7.
Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, et al. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol. 2012;12:140.
Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323(5917):1057–60.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–8.
Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol. 2013;33(5):925–37.
Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. 2010;39(6):491–503.
Colonna L, Lood C, Elkon KB. Beyond apoptosis in lupus. Curr Opin Rheumatol. 2014;26(5):459–66.
Ahamada MM, Jia Y, Wu X. Macrophage polarization and plasticity in systemic lupus erythematosus. Front Immunol. 2021;12: 734008.
Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp (Warsz). 2010;58(5):355–64.
Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, et al. NCBI GEO: archive for functional genomics data sets–10 years on. Nucleic Acids Res. 2011;39(Database issue):D1005-10.
Ko WC, Li L, Young TR, McLean-Mandell RE, Deng AC, Vanguri VK, et al. Gene expression profiling in the skin reveals strong similarities between subacute and chronic cutaneous lupus that are distinct from lupus nephritis. J Invest Dermatol. 2021;141(12):2808–19.
Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, et al. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. Research. 2016. https://doi.org/10.12688/f1000research.9005.2.
da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13.
Fonseka P, Pathan M, Chitti SV, Kang T, Mathivanan S. FunRich enables enrichment analysis of OMICs datasets. J Mol Biol. 2021;433(11): 166747.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–12.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Acknowledgements
Not applicable.
Funding
This research was supported by the Foundation of Jiangsu Health Commission (M2020063) and the China Postdoctoral Science Foundation (2016M591930).
Author information
Authors and Affiliations
Contributions
YZ drafted the manuscript and completed the entire revision process. YPL, QH collected the data. YZ analysed the data. YL acted as the corresponding author, created the study design, and revised the manuscript. DS acted as the cocorresponding author and partly did the study design and the manuscript revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained for renal biopsy from each patient. The research was in compliance with the Declaration of Helsinki and approved by the local ethics committees (XYFY2022-KL245-01).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
The detailed clinical information of the patient.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Liao, Y., Hang, Q. et al. GBP2 acts as a member of the interferon signalling pathway in lupus nephritis. BMC Immunol 23, 44 (2022). https://doi.org/10.1186/s12865-022-00520-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-022-00520-5