
Abstract
Background
Soil salinity is one of the major abiotic stresses that threatens crop growth. Cotton has some degree of salt tolerance, known as the “pioneer crop” of saline-alkali land. Cultivation of cotton is of great significance to the utilization of saline-alkali land and the development of cotton industry. Gossypium hirsutum and G. barbadense, as two major cotton species, are widely cultivated worldwide. However, until recently, the regulatory mechanisms and specific differences of their responses to salt stress have rarely been reported.
Results
In this study, we comprehensively compared the differences in the responses of G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124 to salt stress. The results showed that Hai7124 exhibited better growth than did TM-1 under salt stress, with greater PRO content and antioxidant capability, whereas TM-1 only presented greater K+ content. Transcriptome analysis revealed significant molecular differences between the two cotton species in response to salt stress. The key pathways of TM-1 induced by salt are mainly related to growth and development, such as porphyrin metabolism, DNA replication, ribosome and photosynthesis. Conversely, the key pathways of Hai7124, such as plant hormone signal transduction, MAPK signaling pathway-plant, and phenylpropanoid biosynthesis, are mainly related to plant defense. Further comparative analyses of differentially expressed genes (DEGs) revealed that antioxidant metabolism, abscisic acid (ABA) and jasmonic acid (JA) signalling pathways were more strongly activated in Hai7124, whereas TM-1 was more active in K+ transporter-related genes and ethylene (ETH) signalling pathway. These differences underscore the various molecular strategies adopted by the two cotton species to navigate through salt stress, and Hai7124 responded more strongly to salt stress, which explains the potential reasons for the greater salt tolerance of Hai7124. Finally, we identified 217 potential salt tolerance-related genes, 167 of which overlapped with the confidence intervals of significant SNPs identified in previous genome-wide association studies (GWASs), indicating the high reliability of these genes.
Conclusions
These findings provide new insights into the differences in the regulatory mechanisms of salt tolerance between G. hirsutum and G. barbadense, and identify key candidate genes for salt tolerance molecular breeding in cotton.
Similar content being viewed by others


Introduction
Soil salinity is an important abiotic factor affecting agricultural production and the ecological environment. Currently, approximately 932 million hectares of land are affected by salinity worldwide, of which, approximately 20% of arable land and nearly 33% of irrigated land [1]. Owing to various factors such as low precipitation, high surface evaporation, improper irrigation practices, and industrial pollution, the area of saline-alkali land is increasing at a rate of 1.5 million hectares per year [2, 3]. Soil salinization leads to land degradation and a decrease in agricultural productivity, and can even trigger food security issues [4]. Therefore, how to develop and utilize saline land is one of the significant challenges that scientists are currently dedicated to addressing. Long-term production practices have proven that cultivating salt-tolerant crops is currently the most effective and sustainable method for utilizing saline-alkali land [5], which not only addresses the immediate concerns posed by soil salinization but also offers a pathway toward long-term agricultural resilience and sustainability.
Cotton (Gossypium spp.), as one of the salt-tolerant crops, is referred to as the “pioneer crop” for saline-alkali land. Cultivating cotton is highly important for the exploitation and utilization of saline-alkali land and for the development of the cotton industry. Cotton stands as the primary source of raw materials for the textile industry worldwide, accounting for 35% of the total global fiber consumption [6]. It serves as a major commercial crop in more than 30 of the world’s more than 80 cotton-producing countries [7], with a considerable impact of $600 billion per year on the global national economic value [8]. Despite its inherent salt tolerance and positive contribution to saline-alkali land enhancement, cotton still faces challenges such as low yield, substantial investment requirements, and limited profitability under saline-alkali conditions. In general, salt damage predominantly induces three types of stress in cotton. First, the high concentration of salt in the soil increases the osmotic potential, impeding water absorption by plant roots. Second, when the salt concentration reaches the plant’s threshold, it disrupts ion homeostasis in plant cells, restricting the absorption of other essential nutrients and affecting metabolic pathways, resulting in ion toxicity [9]. Third, osmotic stress and ionic toxicity can also cause oxidative stress, which damages essential biological components in plant cells, including membrane lipids, proteins, and nucleic acids [10].
In response to the deleterious effects induced by salt stress, plants have evolved over time, developing a repertoire of abilities and mechanisms to resist salt damage, including osmotic regulation, ion homeostasis regulation, and reactive oxygen species (ROS) homeostasis regulation [11, 12]. When the soil salt concentration is elevated, the plant first activates its osmotic regulatory mechanism; that is, it reduces the intracellular osmotic potential by absorbing some inorganic ions or synthesizing small organic molecules, thus facilitating the transport of water from the exocytocyte to the intracellular space, thereby stabilizing enzyme activity in the cytoplasm [13]. As the duration of plant exposure to salt damage increases, a significant influx of Na+ enters its cells, leading to ion poisoning. On the one hand, plants absorb excess inorganic salt ions and store them in vacuoles within different organs and tissues in a regionalized manner. On the other hand, plants absorb and accumulate large amounts of K+ from the surrounding environment, decreasing the Na+/ K+ ratio and thereby mitigating the toxic effects of Na+ [14]. Salt stress also triggers oxidative stress in plants, resulting in the accumulation of ROS. Excessive ROS inhibit photosynthesis, respiration and protein synthesis, thus inhibiting plant growth and development. Simultaneously, the regulatory mechanism for restoring ROS homeostasis is activated in response to salt stress, and a series of ROS scavenging-related enzymes or substances are activated or synthesize, ultimately reducing the damage caused by ROS [15]. Furthermore, phytohormone and activities of enzymes related to secondary metabolism also play pivotal roles in regulating the plant responses to salt stress [16].
G. hirsutum and G. barbadense are two important species of cultivated cotton. Despite sharing a common allotetraploid ancestor, the independent domestication process resulted in large differences in plant type, yield, fiber quality, and environmental adaptability [17]. G. hirsutum, recognized for its commendable yield, is the predominant cultivated cotton species, contributing approximately 90% of the global cotton output. Conversely, G. barbadense produces extralong staple or extrafine quality fibers, contributing to less than 5% of the world cotton production [6, 18]. As two main cultivated cotton species, G. hirsutum and G. barbadense play important roles in cotton production, and are also the predominant species grown on saline-alkali lands. It is generally acknowledged that G. barbadense performs better than G. hirsutum under adverse salinity conditions [19]. However, differences in their responses to salt stress have not been reported thus far. Analyzing the degree of salt tolerance differentiation between G. hirsutum and G. barbadense can aid in understanding the mechanism of salt tolerance in cotton; and in identifying the key genes involved, providing genetic resources for molecular breeding of salt tolerance in cotton.
To understand the differences in the salt stress responses and regulatory mechanism of G. hirsutum and G. barbadense. In this study, we comprehensively compared the growth and physiological parameters of G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124 under salt stress, revealing the differences between them at the physiological level. Transcriptome sequencing was subsequently employed to delineate the molecular mechanisms underlying the salt stress responses of both species, clarifying the differences between them at the omics level. Simultaneously, we constructed a protein-protein interaction (PPI) network vis the STRING database and detected key modules. Further comparative analysis of differentially expressed genes (DEGs) between the two cotton species explains the potential reasons for the greater salt tolerance of Hai7124. Finally, the key salt tolerance genes were identified by combining previous genome-wide association studies (GWASs) with quantitative real-time PCR (qRT-PCR), providing a basis for molecular breeding of and molecular mechanism studies on salt tolerance in cotton.
Results
Differences in the growth between G. hirsutum TM-1 and G. barbadense Hai7124 under salt stress
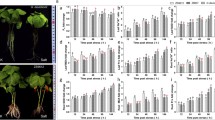
To compare the differences in salt tolerance between the two cotton species, we first observed the morphological changes in TM-1 and Hai7124 under salt stress conditions for one week. As shown in Fig. 1A, TM-1 began to wilt on the first day after 400 mM salt treatment, and the leaves turned slightly yellow on the third day. While Hai7124 did not significantly change on the first day, the leaves began to curl and wilt on the third day, and the leaves began to exhibit some white spots on the fifth day. On the seventh day of salt stress, both cotton species were severely wilted, but that of TM-1 was more serious. Subsequently, we investigated the salt tolerance-related traits of the two cotton species in the treatment group and the control group on the seventh day of salt stress application (Fig. 1B). To eliminate the effects of differences in the growth of the two cotton species, the relative value of each trait was calculated by the ratio of the measurement under stress conditions to the measurement from control plants. The results showed that the differences in salt stress between the two cotton species were mainly manifested in the aerial part of the plant. The relative values of leaf area (LA), plant fresh weight (PFW), and plant dry weight (PDW) of Hai7124 were significantly higher than those of TM-1, by 14.87%, 28.68%, and 15.91%, respectively. However, the relative root shoot ratio (R/S ratio) of TM-1 were significantly greater than that of Hai7124 by 22.93% (Fig. 1C). These results suggest that there are significant differences in the growth of the two cotton species under salt stress, and that Hai7124 has a greater ability to resist salt damage in the early stage of salt stress.

Effect of salt stress on the growth of G. hirsutum TM-1 and G. barbadense Hai7124. A, The change of cotton seedling growth under salt stress for one week; B, Phenotypes of cotton seedlings of G. hirsutum TM-1 and G. barbadense Hai7124 under control and salt stress conditions on the seventh day; C, Relative value of growth-related indexes, including plant height (PH), root length (RL), leaf area (LA), plant fresh weight (PFW), root fresh weight (RFW), plant dry weight (PDW), root dry weight (RDW), and root shoot ratio (R/S). Values are means ± SD (n = 5). Significance level: *p < 0.05, **p < 0.01 (Student’s t-test)
Differences in photosynthesis between G. hirsutum TM-1 and G. barbadense Hai7124 under salt stress
Through morphological observations, we found that the leaves of cotton seedlings were greatly damaged after salt stress, which mainly manifested as leaf curling and wilting. Therefore, we measured the related-indices of cotton seedling leaves. As shown in Fig. 2A and B, the relative electrical conductivity (REC) and relative water content (RWC) did not significantly differ between the two cotton species under normal conditions. However, under salt stress conditions, the REC of Hai7124 was lower than that of TM-1 by 22.96%, and the RWC of Hai7124 was higher than that of TM-1 by 9.80%. Interestingly, through SPAD measurements, we found that the SPAD value of TM-1 was higher than that of Hai7124 by 6.40% under normal conditions. However, the SPAD value of Hai7124 was higher than that of TM-1 by 5.37% under salt stress conditions (Fig. 2C). Furthermore, we measured the gas exchange parameters of the leaves and found that, under normal conditions, the net photosynthetic rate (Pn) and stomatal conductance (Cond) of TM-1 were significantly higher than those of Hai7124, by 29.37% and 14.86%, respectively. However, under salt stress conditions, the Pn, intercellular CO2 concentration (Ci) and transpiration rate (Tr) of Hai7124 were higher than those of TM-1, by 21.36%, 13.83% and 44.28%, respectively. (Fig. 2D-G). These results indicate that Hai7124 maintained greater cellular homeostasis and better photosynthetic capacity under salt stress.

Effects of salt stress on leaf-related indexes and photosynthetic capacity of cotton seedlings. A, Relative electrical conductivity (REC); B, Relative water content (RWC); C, SPAD values; D, Net photosynthetic rate (Pn); D, Stomatal conductance (Cond); E, Intercellular CO2 concentration (Ci); F, Transpiration rate (Tr). Values are means ± SD (n = 5). Significance level: *p < 0.05, **p < 0.01 (Student’s t-test)
Differences in the ion homeostasis and osmotic regulation between G. hirsutum TM-1 and G. barbadense Hai7124 under salt stress
The concentrations of the ratio of Na+/K+ in plants is an important indicator of plant ion homeostasis under salt stress [20]. Therefore, the contents of Na+ and K+ were determined in cotton seedlings in the absence and presence of salt. Obviously, the Na+ content increased dramatically when the cotton seedlings were subjected to salt stress. Interestingly, there was no significant difference in the Na+ content between the two cotton species under either normal or salt stress conditions (Fig. 3A). Conversely, the K+ content decreased when the cotton seedlings were subjected to salt stress. However, the K+ content of TM-1 was higher than that of Hai7124 by 11.66% under salt stress conditions (Fig. 3B), resulting in lower a Na+/K+ ratio in TM-1 (Fig. 3C), which can reduce the damage by excessive Na+ to plants. The PRO content, which protects plant cells from osmotic stress, reflects the stress resistance of plants. As shown in Fig. 3D, we found that the proline (PRO) content of cotton increased dramatically when the seedlings were subjected to salt stress, but the PRO content of Hai7124 was higher than TM-1 by 19.51%. These results suggest that TM-1 has a relatively high K+ content, resulting a lower Na+/K+, which can reduce the damage caused by Na+ to plants under salt stress conditions, whereas Hai7124 has a relatively high proline content, which can reduce the damage caused by osmotic stress to plants under salt stress conditions.

Effects of salt stress on ion accumulation, osmotic balance and antioxidant ability of cotton seedlings. A, Na+ content; B, K+ content; C, Na+/K+ ratio; D, Proline (PRO) content; E, H2O2 content; F, Malondialdehyde (MDA) content; G, Peroxidase (POD) activity; H, Glutathione (GSH) content. Values are means ± SD (n = 5). Significance level: *p < 0.05, **p < 0.01 (Student’s t-test)
Differences in antioxidant ability between G. hirsutum TM-1 and G. barbadense Hai7124 under salt stress
Salt stress causes ion toxicity and osmotic stress, which can lead to the accumulation of high levels of reactive oxygen species (ROS), resulting in ROS toxicity [21]. To detect differences between the excess ROS levels of the two cotton species, the H2O2 contents of TM-1 and Hai7124 were measured in the absence and presence of salt stress. As shown in Fig. 3E, salt stress induced the accumulation of H2O2 in the cotton seedlings. Interestingly, there was a lower H2O2 content in Hai7124 compared to TM-1 under salt stress conditions, significantly decreasing by 25.71%. Malondialdehyde (MDA) is the final decomposition product of membrane lipid peroxidation, and its content can reflect the degree of plant oxidative injury [22]. Similarly, the MDA content also increased when subjected to salt stress. However, MDA content of Hai7124 was lower than that of TM-1 under salt stress conditions, significantly decreasing by 9.05% (Fig. 3F). Furthermore, we measured the peroxidase (POD) activity of the cotton seedlings. As expected, POD activity was significantly induced by salt stress in cotton seedlings, and POD activity in Hai7124 was significantly greater than that in TM-1, by 21.31% (Fig. 3G). Likewise, the glutathione (GSH) content was also measured; similarly, the GSH content was also affected by salt stress, and the GSH content of Hai7124 was significantly greater than that of TM-1, by 29.84% (Fig. 3H). These results are consistent with each other, supporting the conclusion that Hai7124 has relatively strong antioxidant ability, which can reduce the damage caused by excess ROS to plants under salt stress.
Global transcriptome analysis of G. hirsutum TM-1 and G. barbadense Hai7124 under salt stress
To characterize the transcriptional characteristics of these two cotton species, whole-genome expression was evaluated via RNA-seq under normal and salt stress conditions. The results showed that a total of 569.49 million raw reads were initially generated, of which there were approximately 47.46 million reads per sample. After quality control, approximately 567.19 million clean reads remained, with approximately 6.09 Gb per sample, and the Q20 and Q30 percentages of the clean data for all the samples were greater than 95.21% and 90.38%, respectively. The GC content of the clean reads in each sample was more than 43.25%. The clean reads were mapped to the reference genome. We found that 93.81–96.80% of the reads were mapped to those samples, 90.65–93.40% of which were unique mapping reads (Table S1). The PCA results showed that the three replicates of each sampling group clustered well together (Fig. 4A). These results indicated that the clean reads were of high quality for further analysis.

Overview of the transcriptome data in TM-1 and Hai7124. A, PCA of gene expression values of all the samples; B, Number of up- and downregulated DEGs identified in TM-1 and Hai7124, FDR < 0.05 and |log2(fold-change)| >1; C, Expression patterns of the 597 overlapped DEGs between TM-1 and Hai7124, The heatmap presents normalized FPKM expression values; D, Venn diagram of DEGs in TM-1 and Hai7124, including all DEGs and up- and downregulated DEGs
Two different comparison groups were obtained through a comparison of the differentially expressed genes (DEGs) of the same material under normal and salt stress conditions: TM-1 + salt versus TM-1 control (TS vs. TC) and Hai7124 + salt versus Hai7124 control (HS vs. HC). By analyzing the number of DEGs in the two comparison groups, we found that there were significant differences in the response to salt stress between the two cotton species. Specifically, a total of 3420 DEGs were identified in TM-1, namely, 1349 upregulated DEGs and 2071 downregulated DEGs, whereas 2274 DEGs were identified in Hai7124, namely, 1427 upregulated DEGs and 847 downregulated DEGs (Fig. 4B; Table S2; Table S3). An intersection and union analysis of the different DEG sets was performed. There were 597 overlapping DEGs between TM-1 and Hai7124, which were considered to be the core genes whose expression was induced by salt stress. Furthermore, an overlapping set of 334 upregulated and 186 downregulated genes was involved (Fig. 4C). These genes presented similar expression patterns in both TM-1 and Hai7124 (Fig. 4D), suggesting that these genes may have vital effects on the response to salt stress.
Functional characterization of DEGs in G. hirsutum TM-1 and G. barbadense Hai7124
To explore the biological functions of the DEGs, KEGG pathway analysis was performed (Table S4; Table S5). We selected the twenty most enriched pathways from each comparison for further analysis (Fig. 5A and B). Some pathways were shared by the two comparisons, including plant hormone signal transduction, flavone and flavonol biosynthesis, starch and sucrose metabolism and MAPK signaling pathway-plant. Some pathways were uniquely enriched in TM-1 or Hai7124, such as porphyrin metabolism, DNA replication, photosynthesis, valine, leucine and isoleucine degradation and mismatch repair, which were significantly enriched only in TM-1 (q-value < 0.05). However, diterpenoid biosynthesis, carotenoid biosynthesis, plant-pathogen interaction, ubiquinone and other terpenoid-quinone biosynthesis and flavonoid biosynthesis were significantly enriched only in Hai7124 (q-value < 0.05).

The KEGG and GO enrichment analysis of DEGs in TM-1 and Hai7124. Top 20 KEGG enrichment pathways for DEGs in TM-1 (A) and Hai7124 (B); Top 20 significant GO terms enriched in TM-1 (C) and Hai7124 (D), of which there were fewer than 20 significant GO terms in the cellular component and molecular function category
GO functional classification was applied to the DEGs to annotate the functional categories that are active and participate in salt treatment (Table S6; Table S7). For the DEGs of TM-1, the top GO terms in the biological process category were mainly related to the metabolic process, regulation and stress response, such as DNA replication, leaf senescence, cell cycle, response to abiotic stimulus, response to biotic stimulus, response to oxygen-containing compound and so on. In the cellular component category, thylakoid, cell wall and chloroplast were the main GO terms. In the molecular function, the enriched GO terms were mainly involved in DNA-binding transcription factor activity, hydrolase activity and oxidoreductase activity (Fig. 5C). Notably, we found that in Hai7124, the top GO terms in the biological process were mainly related to stress response, including response to chitin, response to endogenous stimulus, response to acid chemical and response to stimulus. In the molecular function category, DEGs were mainly related to nucleus, integral component of plasma membrane and central vacuole. In the molecular function, enriched GO terms were mainly involved transcription factor activity, dioxygenase activity, flavone synthase activity and oxidoreductase activity (Fig. 5D). These results indicate that there are significant molecular differences between the two cotton species in response to salt stress.
PPI network and comparison of the key modules between G. hirsutum TM-1 and G. barbadense Hai7124
Protein-protein interaction (PPI) network and topological attribute analysis can identify important network relationships between proteins from complex biological data. To further understand the functional categories of the DEGs, a PPI network analysis of TM-1 and Hai7124 was conducted. After removing disconnected nodes in the network, we obtained the PPI network of TM-1 with a total of 875 nodes and 2435 edges, involving 1513 DEGs (Fig. S1A; Table S8). And the PP I network of Hai7124 with a total of 388 nodes and 826 edges was obtained, involving 684 DEGs (Fig. S1B; Table S9). Proteins with a high degree of connectivity were considered to play potential key roles in the response of cotton to salt stress. In the PPI network of TM-1, such as GhMCM5 (GH_A13G2142, GH_D13G2127), GhMCM4 (GH_A13G0305, GH_D13G0304), GhRPL5B (GH_A07G1775), GhAUX1 (GH_D13G0173), GhRPL24 (GH_A01G0706, GH_D01G0703), and GhRPL23AB (GH_A10G2350, GH_D10G2446), presented relatively high degree values (≥ 25), which were mainly related to DNA replication, chloroplast protein and metabolism. Likewise, in the PPI network of Hai7124, such as GbMYC2 (GB_D12G1381), GbABI1 (GB_A13G2387, GB_D06G0736), GbMPK3 (GB_A03G0445, GB_A05G1024, GB_D03G1545, GB_D05G1000) GbWRKY33 (GB_A03G0338, GB_A04G1203, GB_D04G1581), GbABI2 (GB_A07G0128, GB_D07G0136, GB_D13G2337), GbPAL1 (GB_D04G1316), GbNCED3 (GB_A01G0351, GB_A08G2779, GB_A13G1899, GB_D01G0370, GB_D08G2765, GB_D13G1842), GbDREB2A (GB_A10G2377, GB_A13G0232, GB_D10G2344), exhibited higher degree values (≥ 15), which were mainly related to transcription, regulation and metabolism.
To further compare the biological functions of the PPI networks of TM-1 and Hai7124, we identified the key modules through MCODE analysis, and performed comparative analysis of the DEGs involved through KEGG and GO enrichment. The results showed there were six key modules obtained from the PPI network of TM-1, including a total of 99 nodes and 717 edges, involving 172 DEGs (Fig. 6A). Three key modules were obtained from the PPI network of Hai7124, including a total of 38 nodes and 109 edges, involving 69 DEGs (Fig. 6B). Furthermore, KEGG enrichment analysis of the key modules showed there were eight pathways significantly enriched in TM-1 (q-value < 0.05), including porphyrin metabolism, DNA replication, ribosome, photosynthesis, fatty acid biosynthesis, flavone and flavonol biosynthesis, circadian rhythm-plant, photosynthesis-antenna proteins. These pathways were mainly related to growth and development. However, there were seven pathways significantly enriched in Hai7124 (q-value < 0.05), including plant hormone signal transduction, circadian rhythm-plant, carotenoid biosynthesis, flavone and flavonol biosynthesis, MAPK signaling pathway-plant, phenylpropanoid biosynthesis, stilbenoid, diarylheptanoid and gingerol biosynthesis. These pathways were mainly related to plant defense (Fig. 6C; Table S10). Similar results were also found in the GO enrichment analysis. Most genes were related to metabolic processes in TM-1, such as DNA replication, organic substance biosynthetic process, DNA metabolic process, cellular biosynthetic process, chlorophyll metabolic process, etc. Conversely, the GO terms were mainly related to plant defense in Hai7124, including response to acid chemical, response to abiotic stimulus, regulation of primary metabolic process, response to chemical, response to osmotic stress, response to hormone, etc. (Fig. 6D; Table S11). These results showed key differences in the salt stress responses of TM-1 and Hai7124. Further manipulation of these genes in stress response pathways may help illustrate their putative regulatory roles in environmental adaptation.

Key modules and enrichment analysis of the modular genes. A, Six key clustering modules of TM-1; B, Three key clustering modules of Hai7124; C, Comparative analysis of KEGG enrichment of the key modular genes between TM-1 and Hai7124; D, Comparative analysis of GO enrichment of the key modular genes between TM-1 and Hai7124
Identification of key salt tolerance genes and pathways
Physiological and biochemical measurements revealed differences in the photosynthetic capacity, K+ content, PRO content and antioxidant ability between TM-1 and Hai7124 (Figs. 2 and 3). On the basis of the transcriptome data, these four types of DEGs were identified in TM-1 and Hai7124, and were considered to be key salt-tolerant genes. There were 26 DEGs associated with photosynthesis, and most of them (22 genes) were significantly downregulated in TM-1. In Hai7124, only four genes were significantly downregulated, and most of the genes did not change significantly (Fig. 7A and D). These results indicate that the photosynthetic capacity of TM-1 is more severely inhibited under salt stress, which is consistent with our photosynthetic measurements. There were four DEGs associated with Na+/H+ exchanger family, and two genes were significantly upregulated in each of TM-1 and Hai7124, with similar expression patterns (Fig. 7B and D). Moreover, 16 DEGs associated with K+ transporter family were identified, seven of which were significantly upregulated and three of which were significantly downregulated in TM-1. In Hai7124, only two genes were significantly upregulated, and six genes were significantly downregulated (Fig. 7B and D). These results indicate that TM-1 has greater K+ transport or absorption capacity under salt stress, which might explain the higher K+ content of TM-1 under salt stress. There were nine DEGs related to proline metabolism, of which five genes were significantly upregulated and three genes were significantly downregulated in TM-1; In Hai7124, three genes were significantly upregulated and only one gene was significantly downregulated (Fig. 7B, D and E). These results explain the salt stress-induced increase in proline content in the two cotton species. Interestingly, the proline content in Hai7124 was greater under salt stress. Furthermore, we identified 55 antioxidation-related DEGs, including POD-encoding genes and genes related to glutathione metabolism, ascorbate metabolism, and flavonoid biosynthesis. Notably, we found that 28 genes were significantly downregulated in TM-1, whereas only 6 genes were significantly upregulated. In contrast, in Hai7124, 30 genes were significantly upregulated, and only four genes were significantly downregulated (Fig. 7C, D, F-H), which may explain the stronger antioxidant capacity of Hai7124.

Identification and comparative analysis of key salt-tolerant genes and pathways between TM-1 and Hai7124. A, Heatmap of gene expression patterns of DEGs related to photosynthesis; B, Heatmap of gene expression patterns of DEGs related to K+ transporter and proline metabolism; C, Heatmap of gene expression patterns of DEGs related to antioxidant; D, Statistics of the number of different types of upregulated and downregulated DEGs identified in TM-1 and Hai7124; E, Proline metabolism pathway; F, Glutathione metabolism pathway; G, Ascorbate metabolism pathway; H, Flavonoid biosynthesis pathway
Phytohormone signaling plays an important regulatory role in the plant response to abiotic stress. Meanwhile, the salt stress-induced pathways in both TM-1 and Hai7124 involved “plant hormone signal transduction” (Fig. 5A and B). Therefore, we also mined genes related to plant hormone signal transduction. As a result, a total of 108 DEGs were identified, which involved abscisic acid (ABA), auxin (AUX), brassinosteroid (BR), ethylene (ETH) and jasmonic acid (JA) signaling pathways. There were 24 DEGs related to ABA signaling, most of which (15 genes) were significantly upregulated in Hai7124. In TM-1, only six genes were significantly upregulated, and most of the genes did not change significantly (Fig. 8A, D and E). There were 41 DEGs related to AUX signaling, among which 16 genes in TM-1 were upregulated and 13 genes were downregulated. In Hai7124, only seven genes were significantly upregulated and 15 genes were significantly downregulated (Fig. 8B, D and F). There were 15 DEGs associated with BR signaling, among which six genes in TM-1 were upregulated and six genes were downregulated. In Hai7124, only four genes were significantly upregulated, and the other genes did not change significantly (Fig. 8C, D and G). Fifteen genes were related to ETH signaling, of which ten and six genes were upregulated in TM-1 and Hai7124, respectively, and no significant changes were detected in the other genes (Fig. 8C, D and H). There were 13 genes associated with JA signaling, of which two genes were significantly downregulated in TM-1 and only one gene was significantly upregulated. However, ten genes in Hai7124 were significantly upregulated (Fig. 8C, D and I). These results indicate that there are significant differences in plant hormone signal transduction between the two cotton species under salt stress. Hai7124 mainly responds to salt stress by upregulating ABA and JA-related genes, while TM-1 mainly responds to salt stress by ETH-related genes.

Identification and comparative analysis of plant hormone-related genes and pathways between TM-1 and Hai7124. A, Heatmap of gene expression patterns of DEGs related to abscisci acid; B, Heatmap of gene expression patterns of DEGs related to auxin; C, Heatmap of gene expression patterns of DEGs related to brassinosteroid, ethylene, jasmonic acid; D, Statistics of the number of different types of upregulated and downregulated DEGs identified in TM-1 and Hai7124; E, Abscisic acid signaling pathway; F, Auxin signaling pathway; G, Brassinosteroid signaling pathway; H, Ethylene signaling pathway; I, Jasmonic acid signaling pathway
Verification of key salt tolerance genes via previous GWAS results
To determine the accuracy of salt tolerance genes identified in this study, seven previous GWAS studies related to salt tolerance in cotton were reviewed [23,24,25,26,27,28,29]. These GWAS studies utilized populations of six G. hirsutum accessions and one G. barbadense accessions with population sizes ranging from 217 [27] to 713 [26]. We then collected the most significant SNPs reported in each GWAS study and performed a comparative analysis with the salt tolerance genes we identified. The results revealed a total of 841 significant SNPs related to salt tolerance in cotton (Fig. 9; Table S12). The distribution of these SNPs on each chromosome was different. Most of the significant SNPs were located on D08 (46), whereas only six SNPs were located on A08. In general, the LD decay of the GWAS population could be used as a confidence interval for significant SNPs in the genome. The LD decay of these seven GWAS studies varied from 200 to 869 kb, with an average LD decay of 500 kb, so confidence intervals of significant SNPs were identified within 500 kb upstream or downstream of peak SNPs. To determine the accuracy of the salt tolerance genes, the above 217 genes were compared with the confidence intervals of 841 significant SNPs. The results showed 167 genes overlapped confidence interval of the significant SNPs. The number of overlapping genes on each chromosome varied, ranging from 15 for A05 to 2 genes for D03. These results indicate that 167 of the 217 salt tolerance genes identified in this study overlap with previous GWAS reports, indicating their high reliability.

Comparative analysis of salt-tolerance genes we mined with previous GWAS results. The left border of each chromosome represents the distribution of SNPs associated with salt tolerance on the chromosome, and the right border represents the distribution of salt-tolerance genes we mined
Validation of key salt tolerance genes via qRT-PCR
On the basis of functional and comparative analysis with previous GWAS results, we selected ten salt tolerance genes in TM-1 and Hai7124 to monitor their expression levels at different time points under salt stress (Fig. 10A). GH_A08G0880 is mainly related to the potassium transporter, and its expression trend was upregulated upon salt induction, peaking at 12 h, after which it decreased. Notably, we observed higher expression levels of GH_A09G0318 in TM-1 than in Hai7124 (Fig. 10B). The expression trend of GH_D06G1914 was continuously downregulated by salt induction, which was mainly related to photosynthesis. However, there was a greater expression level in Hai7124 than in TM-1 (Fig. 10C). GH_A09G1908 and GH_A07G0814 are related to peroxidase and glutathione metabolism, respectively. Their expression trends were similar, namely, salt-induced upregulated expression, peaking at 6–12 h, and then decreasing. However, there was a higher expression level in Hai7124 than in TM-1 (Fig. 10D-E). GH_A04G1046, which is associated with proline metabolism, was continuously upregulated by salt induction. The expression level of GH_A04G1046 was higher in Hai7124 upon salt stress (Fig. 10F). Furthermore, the expression of five plant hormone signaling-related genes was also detected. GH_A05G0933 and GH_D06G0153 were related to ABA and BR signaling, respectively. Their expression trends were similar, namely, salt-induced upregulated expression, peaking at 12 h, and then decreasing (Fig. 10G and J). The expression of GH_A02G0373 and GH_A01G0218, which are related to AUX and JA signaling, respectively, fluctuated in response to salt stress at different time points (Fig. 10H and I). However, the expression of GH_A09G1664, which is associated with ETH signaling, was continuously upregulated by salt induction (Fig. 10K). Notably, with the exception of GH_D06G0153, the expression levels of these genes in Hai7124 were higher than those in TM-1, suggesting their potentially pivotal roles in improving plant salt tolerance.

Validation of the expression of ten salt-tolerant genes via qRT-PCR. A, Functional annotation and classification of ten genes selected; B-K, Expression levels of ten genes at different time points via qRT–PCR. Significance level: *p < 0.05, **p < 0.01 (Student’s t-test)
Discussion
Cotton growth is affected by various abiotic stresses, among which soil salinity is one of the major factors restricting cotton productivity. Different cotton germplasm resources have been shown to adapt to salt stress in different ways [30, 31], and it is important to study and compare the differences in their responses to salt stress for the breeding of salt-tolerant cotton. When plants are exposed to salt stress, their growth and development are inhibited, resulting in symptoms such as stunted growth and withered leaf margins, ultimately leading to a reduction in biomass [10]. Similarly, in this study, we observed that G. hirsutum TM-1 and G. barbadense Hai7124 also suffered severe growth inhibition under salt stress. However, the two cotton species presented different phenotypic changes during salt stress, with Hai7124 being more salt-tolerant, mainly manifested in the above-ground parts, especially on the leaves (Fig. 1A-C), which prompted us to further explore.
Photosynthesis provides plants with essential substances and energy, plays a crucial role in plant growth and development. Under the adversity environment, the growth and development of plants are severely inhibited, which ultimately feeds back to the photosynthesis of plants, therefore, the strength of photosynthetic capacity is an important indicator of plant resistance to adversity stress [32, 33]. Interestingly, in our study, the photosynthetic capacity of TM-1 was greater than that of Hai7124 under normal conditions; however, under salt stress conditions, both exhibited inhibited photosynthesis to a significant extent, but the photosynthetic capacity of Hai7124 was greater than that of TM-1 (Fig. 2C-G). These results suggest that Hai7124 may have a stronger photosynthetic protection mechanism under adverse salt conditions. In fact, decreased photosynthetic capacity is a form of regulation that plants produce in response to adverse environments to limit the metabolic costs required for photosynthetic mechanisms under stress conditions [34, 35]. However, prolonged exposure of plants to saline environments can result in osmotic stress as well as excessive accumulation of Na+ or ROS, resulting in cytoplasmic membrane damage or ion toxicity, and severely inhibiting plant photosynthesis [36]. Generally, plants resist the damage caused by salt stress mainly through ion homeostasis regulation, osmotic regulation and ROS homeostasis regulation [11, 12, 37,38,39]. Depolarisation of the plasma membrane resulting from the influx of Na+ into the cell causes a continuous efflux of K+ from the cell under salinity stress [3]. Therefore, intracellular Na+/K+ is an important indicator of plant ion homeostasis under salt stress [40]. Our results showed that TM-1 had better ion homeostasis under salt stress (Fig. 3A-C). PRO is of great significance for maintaining the osmotic balance of plant cells under environmental stress. The higher level of PRO can protect cotton from osmotic pressure under salt stress [28]. Moreover, salt stress can lead to the accumulation of ROS, and a certain amount of ROS can be used as a signal factor to stimulate the salt stress tolerance pathway of plants [10]. However, excessive ROS cause cytoplasmic membrane damage and metabolic dysfunction [24, 29,30,31,32]. Our results suggested that Ha7124 had stronger osmotic regulation and antioxidant capacity under salt stress (Fig. 3D-H). These results reveal physiological differences between the two cotton species in response to salt stress, and the diversity of salt tolerance patterns may be the reason for the greater salt tolerance of Hai7124 than TM-1.
When plants are subjected to salt stress, salt can trigger global changes in the metabolic pathways of plants, providing insight for the exploration of key regulatory mechanisms [2]. From the current results, we supposed that some pathways, such as plant hormone signal transduction, flavone and flavonol biosynthesis, starch and sucrose metabolism and MAPK signaling pathway-plant, flavonoid biosynthesis were active when cotton suffers from salt stress (Fig. 5A and B). Plant hormone signal transduction, MAPK signaling pathways and favonoid biosynthesis are regarded as basic response pathways in cotton, and these pathways are triggered both in TM-1 and Hai7124. On the basis of these findings, we can conclude that there is a set of basic and broad-spectrum metabolic pathways that respond to stress, which endows plants with general resistance. These results are consistent with those of several previous reports [22, 41, 42]. In addition, we detected differences in the resistance levels of TM-1 and Hai7124, which are linked to specific response mechanisms and countermeasures (Fig. 5A and B). Pathways related to plant growth and development, such as porphyrin metabolism, DNA replication, ribosome, photosynthesis, were mainly enriched in TM-1. However, the pathways involved in plant resistance, such as plant-pathogen interaction, diterpenoid biosynthesis, carotenoid biosynthesis, and phenylpropanoid biosynthesis, were mainly enriched in Hai7124. These differences underscore the various molecular strategies adopted by the two cotton species to manage salt stress, and Hai7124 may exhibit earlier and stronger responses to salt stress.
The identification of DEGs involved in the key metabolic processes or pathways can provide detailed insights into the differences in salt tolerance between these two cotton species. We found that most of the genes (22 genes) in TM-1 were significantly downregulated in the photosynthetic pathway (Fig. 7A and D). Some genes, such as PsaK, PsaL and PsaN in photosystem I, and PsbQ, Psb27 and PsbS in photosystem II, play important roles in the photosynthetic system [32, 33], but are severely inhibited in TM-1 under salt stress. These results were consistent with our previous measurements and further indicate that Hai7124 has a stronger salt tolerance. The Na+/H+ exchanger family (NHX) can increase Na+ efflux or compartmentalize Na+ in the vacuole [9, 41]. In our study, two NHXs were significantly upregulated in each of TM-1 and Hai7124, suggesting enhanced vesicular sequestration and efflux capabilities, which might attenuate cell damage due to salt-induced stress [43]. Moreover, similar expression patterns may explain the lack of difference in the Na+ contents of the two cotton species. Importantly, more K+ transport-related genes, including HKT, KAT and KT, were upregulated in TM-1 (Fig. 7B and D). These genes mainly regulate K+ transport and uptake, which may enable TM-1 to maintain the K+ supply under salt exposure [9, 44], explaining the higher K+ content of TM-1 under salt stress. PRO is highly important for maintaining the osmotic balance of plant cells under environmental stress [45]. We found that both TM-1 and Hai7124 were activated under salt stress (Fig. 7B, D-E), and the key genes involved in PRO synthesis, P5CS and PRODH [3, 9], were upregulated, which explain the increase in PRO content in the two cotton species under salt stress. Interestingly, the PRO content in Hai7124 under salt stress was greater than that of TM-1, which may be caused by the different protein activities encoded by related genes.
Moreover, salt stress can lead to the accumulation of ROS, excessive ROS cause cytoplasmic membrane damage and metabolic dysfunction [10, 32, 36]. In general, plants need to produce antioxidant defense systems, such as antioxidant enzymes or substances, in response to excessive ROS [37,38,39]. On the one hand, antioxidant enzymes can react directly with excessive ROS to reduce the damage caused by ROS to plants. In our study, two POD-encoding genes were upregulated in Hai7124, whereas only one was upregulated in TM-1 (Fig. 7C, D and H), which is consistent with our findings that Hai7124 has greater POD activity. On the other hand, the nonenzymatic antioxidants glutathione and ascorbate can react with membrane lipid peroxidation products to terminate their peroxidation and scavenge ROS through the metabolic cycle [9, 46]. Our results revealed that genes related to the synthesis of antioxidant substances, including the key genes involved in GSH synthesis, GST, GPX and PRDX6, and the key genes involved in ascorbic acid synthesis, ASO, GAE and AKR1B [34, 38], were mostly upregulated in Hai7124; thus more antioxidants may be produced to reduce the damage caused by ROS in plants. In addition, flavonoids, as secondary metabolites, have a strong antioxidant capacity [11, 12, 34]. Some genes, such as FLS, CYP75A, CYP73A, and F3H, play important roles in flavonoid biosynthesis [32, 33, 35], and they are highly expressed in Hai7124, implying that Hai7124 may synthesize more secondary metabolites, resulting in greater antioxidant capacity. These results are consistent with each other, underscoring the reliability of the current findings.
Phytohormone are important signaling molecules that are produced in plants at very low concentrations, but are involved in a wide range of processes, including flower, stem, and leaf formation, fruit ripening, and the response to biotic or abiotic stresses [11]. In this study, many genes involved in hormonal signaling pathways, including ABA, AUX, BR, ETH and JA, were found to be involved in the response of cotton to salt. Among them, ABA, as the core stress response hormone, plays an irreplaceable role in salt stress defense [47]. In our study, we found that the carotenoid biosynthesis pathway in Hai7124 was significantly enriched (Fig. 5B), and the ABA signaling pathway was more active in Hai7124 under salt conditions (Fig. 8A, D-E). On the one hand, ABA accumulates in plant cells under salt stress and promotes the expression of salt-responsive genes, which are strongly related to salt tolerance mechanisms in plants [28]. Exogenous ABA and the overexpression of genes involved in ABA biosynthesis alleviate the adverse effects of salt by increasing osmotic and ion regulation, antioxidant accumulation, and flavonoid metabolism [48]. On the other hand, the ABA signaling pathway can interact with other hormone signals to regulate plant responses to external stress [49]. For instance, low salt stress can induce a small amount of ABA, activate AUX signaling and the production of lateral roots, whereas high salt stress leads to excessive accumulation of ABA, disrupting auxin distribution and lateral roots development [47]. In this study, auxin signaling was induced in both TM-1 and Hai7124, but most of the genes were downregulated (Fig. 8B, D and F), possibly because ABA induced by high salt stress disrupted AUX signaling in both cotton species [50], among which TM-1 disrupted more seriously. Similarly, ABA also inhibits root growth by promoting ETH signaling [51], which upregulates the expression of peroxidase and prevents ROS damage in plants [52]. In our study, both TM-1 and Hai7124 induced the expression of genes related to ETH signaling, but the ETH signaling pathway was more active in TM-1 under salt conditions (Fig. 8C, D and H). Moreover, BR are widespread hormones in plants, that play key roles in cell differentiation, plant growth and abiotic stress response [47]. Exogenous BR on leaves and roots can reduce the degree of damage caused by salt stress in plants [53]. In our study, TM-1 presented both upregulated and downregulated genes related to BR signaling, whereas Hai7124 only presented upregulated genes related to BR signaling (Fig. 8C, D and G), indicating that the two respond to salt stress through different BR signaling patterns. JA enhances salt tolerance in plants mainly through ROS scavenging pathways [48]. The JA signaling pathway was more active in Hai7124 under salt conditions (Fig. 8C, D and I), which explained the increased antioxidant capacity of Hai7124. These results indicate differences in hormone signaling between TM-1 and Hai7124 in response to salt stress. These genes or pathways provide an important reference for further exploration of the molecular mechanism of salt tolerance in cotton. Moreover, the key salt-tolerant genes we mined, especially those overlapping with previous GWAS results (Fig. 9), provide important resources for cotton salt-tolerant molecular breeding [23,24,25,26,27,28,29]. Further manipulation of these genes in key response pathways may help illustrate their putative regulatory role in environmental adaptation.
Conclusion
In this study, phenotypic, physiological and transcriptomic analyses were performed to compare differences in the salt stress response between G. hirsutum TM-1 and G. barbadense Hai7124. Phenotypically, salt stress severely inhibited the growth of both cotton species, with Hai7124 exhibiting better growth characteristics, especially on the leaves. Physiologically, compared with TM-1, Hai7124 presented superior osmotic regulation and antioxidant capabilities, whereas TM-1 only presented greater ion homeostasis regulation than did Hai7124 under salt stress. Further transcriptome analysis revealed that the molecular response of TM-1 under salt stress was mainly related to growth and development, whereas that of Hai7124 was mainly related to plant defense, implying that Hai7124 can respond more strongly to salt stress. The comparative analysis of DEGs between the two cotton species, which are involved in key pathways, explains the potential reasons for the greater salt tolerance of Hai7124. Finally, we identified 217 potential salt-tolerant genes, 167 of which overlapped with the confidence intervals of significant SNPs from previous GWAS studies. These results provide new insights into the differences in the regulatory mechanisms of salt tolerance between G. hirsutum and G. barbadense, and identify key candidate genes for salt tolerance molecular breeding in cotton.
Materials and methods
Plant materials, culture conditions and growth measurements
The seeds of G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124 were provided by Nanjing Agricultural University, Nanjing City, Jiangsu Province, China. All the seeds were surface sterilized with 75% ethanol for 1 min and 2% sodium hypochlorite for 5 min, followed by washing three times with sterile distilled water (3 min each). Surface-sterilized seeds were sown in pots filled with peat soil: vermiculite (1:1) and cultured in the greenhouse (25 °C, 14/10 h light/dark, 60% relative humidity). When growing to the two-leaf stage, seedlings with identical growth sizes were selected and divided into two groups: one was root-irrigated with 70 mL of 400 mM NaCl, and the other was treated with an equal volume of water as the control. The morphological characteristics of the two cotton species were observed and recorded from 0 to 7 days of salt stress application. After one week of salt stress, five seedlings were randomly selected for each treatment, and the morphological traits, i.e., plant height (PH, cm), root length (RL, cm), leaf area (LA, cm2), plant fresh weight (PFW, g), plant dry weight (PDW, g), root fresh weight (RFW, g), and root dry weight (RDW) were measured. The root to shoot ratio (R/S) was calculated as the ratio of RDW to PDW.
Measurements of SPAD, RWC, REC and photosynthetic parameters of leaves
The SPAD values were determined by SPAD meter (Minolta Camera Co., Osaka, Japan), representing the chlorophyll contents of the leaves. The leaves from the same part of each plant were collected, weighed, and recorded as the fresh weight (FW). The leaves were then soaked in deionized water for 24 h and removed to wipe the surface moisture, the leaves were weighed and recorded as saturated fresh weight (SFW). Finally, the leaves were placed at 105 °C for 30 min, then dried at 80 °C to constant weight, and dry weight (DW) was measured after cooling. The relative water content was calculated as follows: relative water content (RWC, %) = (FW - DW) / (SFW - DW) × 100% as described previously [54]. Likewise, the leaves from the same part of the plant were collected for electrical conductivity determination. The leaves were cut into pieces and soaked for 24 h in centrifuge tubes filled with 20 mL deionized water at room temperature. A conductivity meter was used to measure the electrical conductivity before (E1) and after (E2) heating the samples in a boiling water bath for 30 min (the samples were cooled to room temperature before taking the measurement). Thereafter, the relative electrical conductivity was calculated as follows: relative electrical conductivity (REC, %) = (E1/E2) × 100% [2]. The photosynthetic parameters were measured from 8:50 am to 11:00 am, two leaves at the top of each seedling were chosen as test sites. Li-6800 photosynthetic apparatus ((Li-COR, Lincoln, NE, USA)) was used to measure photosynthetic parameters, including the Net photosynthetic rate (Pn), Stomatal conductance (Cond), Intercellular CO2 concentration (Ci) and Transpiration rate (Tr). The self-set light intensity of the LED was controlled at 800 µmol m− 2 s− 1 and the temperature was 25 ± °C.
Na+ and K+ content determination
The Na+ and K+ contents were measured via the method reported previously [55]. Briefly, dried samples (0.5 g accurately measured to 0.0001 g) were weighed into the inner vial of the Teflon digestion tank and soaked in 5 mL of nitric acid. A thermostatic drying oven was then used to dry the samples at 80 and 120 °C for 2 h, and at 160 °C for 4 h. The samples were cooled to room temperature, opened under a fume chamber, and heated until the acid evaporated. The obtained digestive liquid was transferred into a 25 mL volumetric flask, and a small amount of nitric acid solution (1%) was used to rinse the residual samples (3 times) from vessels (the inner jar and its cover) into the volumetric flask. The sample was then scaled up using 1% nitric acid forming the test liquid. The ion content was determined using an inductively coupled plasma-emission spectrometer (Thermo Fischer Scientific, Waltham, MA, USA). The Na+/K+ ratio was calculated in line with the Na+ and K+ contents.
Biochemical index determination
The contents of proline (PRO), hydrogen peroxide (H2O2), malondialdehyde (MDA), glutathione (GSH) and peroxidase (POD) activity were measured via commercially available kits (Sangon Biotech Co., Ltd., Shanghai, China). The PRO content was determined based on reactivity with an acidic ninhydrin solution. Briefly, the sample was homogenized in aqueous sulfosalicylic acid and centrifuged to obtain the supernatant, which was then mixed with acidic ninhydrin solution to form a red product with characteristic absorption at 520 nm. The PRO content was determine based on a standard curve with pure proline at known concentrations. The H2O2 content was determined based on H2O2 bound with titanium sulfate to form a complex. Briefly, the sample was homogenized in acetone and centrifuged to obtain the supernatant, and titanium sulfate was added to form a yellow titanium peroxide complex with characteristic absorption at 415 nm. The MDA content was determined based on thiobarbituric acid (TBA) reactivity. Briefly, the sample was homogenized in trichloroacetic acid and centrifuged d to obtain the supernatant, and TBA was added to form the brown-red trimethadione, which has a maximum absorption wavelength of 532 nm. Finally, the difference in absorbance between 532 nm and 450 nm and 600 nm was used to calculate the content of MDA. The contents of GSH were determined based on the reaction with 5,5’-dithiobis-2-nitrobenzoic acid (DTNB). Briefly, the sample was homogenized in aqueous sulfosalicylic acid and centrifuged to obtain the supernatant, which is then mixed with DTNB to form a yellow product, which has a maximum absorption wavelength of 412 nm. For POD activity, the sample was homogenized in phosphate buffer solution (PBS, pH 7.8) and centrifuged to obtain the supernatant. POD activity was calculated based on the change of absorbance at 470 nm resulting from the catalysis of H2O2. One unit was defined as the amount of enzyme that catalyzes the reaction and generates 1 µg of substrate from 1.0 g fresh tissues in the reaction system at 37 °C.
RNA-Seq and differential expression analysis
The cotton leaves of the salt group and control group were collected at 24 h. Three replicates were randomly collected for each treatment, and immediately frozen in liquid nitrogen and stored at -80 °C. Total RNA was extracted via an RNA Plant Kit (Tanigen) following the manufacturer’s recommendations. Total RNA was quantified via a Qubit (Thermo Fisher Scientific) and the RNA quality was checked via a Tapestation 2200 (Agilent Technologies, Santa Clara, CA, USA). The mRNA was fragmented and transcribed into cDNA to construct libraries. The cDNA libraries were then sequenced using Illumina NovaSeq 6000 by Gene Denovo Biotechnology Co., Ltd (Guangzhou, China). The sequences were deposited at the Sequence Read Archive of the National Center for Biotechnology (https://www.ncbi.nlm.nih.gov/sra) under the BioProject number PRJNA1099770.
Clean raw data were obtained through quality control via Fastp (version 0.19.4) [56]. The paired-end clean reads were subsequently aligned against the reference genome sequence of G. hirsutum cv. TM-1 and G. barbadense cv. Hai7124 [57] via Hisat2 (version 2.2.1) [58]. The mapped reads of each sample were assembled via StringTie (version 2.1.4) software [59], and gene expression abundance of each sample was calculated via RSEM [60]. Finally, the fragments per kilobase of exon per million fragments mapped (FPKM) value for each gene were obtained, which was used as the measure of the gene expression level. The principal component analysis (PCA) and Pearson’s correlation coefficient (PCC) analysis were performed using Prcomp and Cor functions in R (version 3.6.4). The differentially expressed genes (DEGs) were identified using DESeq2 (version 1.26.0) [61] with the thresholds of 2-fold expression changes [|log2(fold-change) |>1] and false discovery rate (FDR) < 0.05. Subsequently, all DEGs were mapped to Gene Ontology (GO) terms in the GO database (http://www.geneontology.org), and gene numbers were calculated for every term. Significantly enriched GO terms (q-value < 0.05) in DEGs compared with the genome background were defined by hypergeometric testing. Pathway enrichment analysis was also performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Pathways with q-value < 0.05 were defined as significantly enriched pathways in DEGs.
Protein-protein interaction network construction
The protein–protein interaction (PPI) network of DEGs was predicted via STRING [62] (https://string-db.org/), in which interactions with a combined score over 0.7 were considered statistically significant. Cytoscape (http://www.cytoscape.org) (version 3.9.0) [63] was used to visualize this PPI network. Cytoscape’s plug-in molecular complex detection technology (MCODE) was used to analyze key functional modules [64]. Set the selection criteria as: K-core = 2, degree cutoff = 2, max depth = 100, and node score cutoff = 0.2. Then the KEGG and GO analysis of the involved modular genes were performed.
Verification by previous GWAS studies
To determine the accuracy of the identified salt tolerance genes identified in this study, seven previous GWAS studies related to salt tolerance in cotton were reviewed [23,24,25,26,27,28,29]. We subsequently collected the reported peak SNPs (the most significant SNPs) and associated traits from each GWAS study. The physical positions of these SNPs are based on the common reference genome sequence of G. hirsutum cv. TM-1 [57]. Finally, we compared whether the locations of the salt tolerance genes overlapped with the confidence intervals of the significant SNPs to determine their accuracy.
Quantitative real-time PCR (qRT-PCR)
Total RNA was reverse transcribed into cDNA via a HiScript II Reverse Transcriptase Kit (Genesand, Beijing, China). qRT-PCR reactions were performed via a StepOnePlusTM Real-Time PCR System (Thermo Fisher Scientific) in a 20 µl volume containing 10 µl of 2 ×GS AntiQ qPCR SYBR Master Mix, 0.4 µl of ROX Reference Dye 1 (High), 2 µl of cDNA template, and 0.8 µl each of forward and reverse primer (Genesand, Beijing, China). The PCR conditions were as follows: predenaturation at 95 °C for 1 min followed by 40 amplification cycles of 20 s at 95 °C and 30 s at 60 °C. After the cycle was complete, melting curve analysis was performed at 60–95 °C to verify the specificity of the amplicon for each primer pair. qRT-PCR was carried out with three biological replicates, and each replicate included three technical repeats. The values from triplicate reactions were averaged, the cotton actin gene was used as an internal reference, and the Ct values of every gene were determined and compared with the 2−ΔΔCt method [65]. The gene-specific primers used in this analysis are listed in Table S13.
Statistical analysis
All the data were organized and analyzed via Microsoft Excel and GraphPad Prism version 8.2.1 (GraphPad Software, Boston, Massachusetts USA, www.graphpad.com). The results are expressed as the mean values ± standard deviations (SD) of all the replications. The statistical significance of differences between treatments was assessed by Student’s t test. All significant values are indicated as ∗, p < 0.05, or ∗∗, p < 0.01.
Data availability
The raw RNA-seq data are available in the NCBI database (https://www.ncbi.nlm.nih.gov/geo/) under accession number PRJNA1099770. All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Dong Y, Chen R, Petropoulos E, Yu B, Zhang J, Lin X, Gao M, Feng Y. Interactive effects of salinity and SOM on the ecoenzymatic activities across coastal soils subjected to a saline gradient. Geoderma. 2022;406:115519.
Feng L, Li Q, Zhou D, Jia M, Liu Z, Hou Z, Ren Q, Ji S, Sang S, Lu S, Yu J. B. subtilis CNBG-PGPR-1 induces methionine to regulate ethylene pathway and ROS scavenging for improving salt tolerance of tomato. Plant J. 2024;117(1):193–211.
Munns R, Tester M. Mechanisms of salinity tolerance. Annu Rev Plant Biol. 2008;59:651–81.
van Zelm E, Zhang Y, Testerink C. Salt tolerance mechanisms of plants. Annu Rev Plant Biol 71; 403–33.
Cao Y, Song H, Zhang L. New insight into plant saline-alkali tolerance mechanisms and application to breeding. Int J Mol Sci. 2022;23:16048.
Chen Z, Scheffler BE, Dennis E, Triplett BA, Zhang T, Guo W, Chen X, Stelly DM, Rabinowicz PD, Town CD, Arioli T, Brubaker C, Cantrell RG, Lacape JM, Ulloa M, Chee P, Gingle AR, Haigler CH, Percy R, Saha S, Wilkins T, Wright RJ, Van Deynze A, Zhu Y, Yu S, Abdurakhmonov I, Katageri I, Kumar PA, Mehboob-Ur-Rahman, Zafar Y, Yu JZ, Kohel RJ, Wendel JF, Paterson AH. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007;145(4):1303–10.
Abdelraheem A, Esmaeili N, O’connell M, Zhang J. Progress and perspective on drought and salt stress tolerance in cotton. Ind Crops Prod. 2019;130:118–29.
Ashraf J, Zuo D, Wang Q, Malik W, Zhang Y, Abid MA, Cheng H, Yang Q, Song G. Recent insights into cotton functional genomics: progress and future perspectives. Plant Biotechnol J. 2018;16(3):699–713.
Yang Y, Guo Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018;217(2):523–39.
Deinlein U, Stephan AB, Horie T, Luo W, Xu G, Schroeder JI. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014;19(6):371–9.
Fang S, Hou X, Liang X. Response mechanisms of plants under saline-alkali stress. Front Plant Sci. 2021;12:667458.
Zhou H, Shi H, Yang Y, Feng X, Chen X, Xiao F, Lin H, Guo Y. Insights into plant salt stress signaling and tolerance. J Genet Genomics. 2024;51(1):16–34.
Chérel I, Lefoulon C, Boeglin M, Sentenac H. Molecular mechanisms involved in plant adaptation to low K+ availability. J Exp Bot. 2014;65(3):833–48.
Yang Y, Wu Y, Ma L, Yang Z, Dong Q, Li Q, Ni X, Kudla J, Song C, Guo Y. The Ca2+ sensor SCaBP3/CBL7 modulates plasma membrane H+-ATPase activity and promotes alkali tolerance in Arabidopsis. Plant Cell. 2019;31(6):1367–84.
Liu C, Lin JZ, Wang Y, Tian Y, Zheng HP, Zhou ZK, Zhou YB, Tang XD, Zhao XH, Wu T, Xu SL, Tang DY, Zuo ZC, He H, Bai LY, Yang YZ, Liu XM. The protein phosphatase PC1 dephosphorylates and deactivates CatC to negatively regulate H2O2 homeostasis and salt tolerance in rice. Plant Cell. 2023;35(9):3604–25.
Chen MX, Lu CC, Sun PC, Nie YX, Tian Y, Hu QJ, Das D, Hou XX, Gao B, Chen X, Liu SX, Zheng CC, Zhao XY, Dai L, Zhang J, Liu YG. Comprehensive transcriptome and proteome analyses reveal a novel sodium chloride responsive gene network in maize seed tissues during germination. Plant Cell Environ. 2021;44(1):88–101.
He L, Han Z, Zang Y, Dai F, Chen J, Jin S, Huang C, Cheng Y, Zhang J, Xu B, Qi G, Cao Y, Yan S, Xuan L, Zhang T, Si Z, Hu Y. Advanced genes expression pattern greatly contributes to divergence in Verticillium wilt resistance between Gossypium barbadense and gossupium hirsutum. Front Plant Sci. 20221; 13: 979585.
Wendel JF, Brubaker CL, Seelanan T. The origin and evolution of Gossypium. In: Stewart JM, Oosterhuis DM, Heitholt JJ, Mauney J R, editors. Physiology of cotton. Dordrecht: Springer; 2010. pp. 1–18.
Maryum Z, Luqman T, Nadeem S, Khan SMUD, Wang B, Ditta A, Khan MKR. An overview of salinity stress, mechanism of salinity tolerance and strategies for its management in cotton. Front Plant Sci. 2022;13:907937.
Garriga M, Raddatz N, Véry AA, Sentenac H, Rubio-Meléndez ME, González W, Dreyer I. Cloning and functional characterization of HKT1 and AKT1 genes of Fragaria spp.-relationship to plant response to salt stress. J Plant Physiol. 2017;210:9–17.
Li F, Zhang H, Zhao H, Gao T, Song A, Jiang J, Chen F, Chen S. Chrysanthemum CmHSFA4 gene positively regulates salt stress tolerance in transgenic chrysanthemum. Plant Biotechnol J. 2018;16(7):1311–21.
Li J, Ma M, Sun Y, Lu P, Shi H, Guo Z, Zhu H. Comparative physiological and transcriptome profiles uncover salt tolerance mechanisms in Alfalfa. Front Plant Sci. 2022;13:931619.
Abdelraheem A, Kuraparthy V, Hinze L, Stelly D, Wedegaertner T, Zhang J. Genome-wide association study for tolerance to drought and salt tolerance and resistance to thrips at the seedling growth stage in US Upland cotton. Ind Crops Prod. 2021;169:113645.
Cai C, Zhu G, Zhang T, Guo W. High-density 80 K SNP array is a powerful tool for genotyping G. Hirsutum accessions and genome analysis. BMC Genomics. 2017;18(1):654.
Su X, Zhu G, Song X, Xu H, Li W, Ning X, Chen Q, Guo W. Genome-wide association analysis reveals loci and candidate genes involved in fiber quality traits in sea island cotton (Gossypium barbadense). BMC Plant Biol. 2020;20(1):289.
Sun Z, Li H, Zhang Y, Li Z, Ke H, Wu L, Zhang G, Wang X, Ma Z. Identification of SNPs and candidate genes associated with salt tolerance at the seedling stage in cotton (Gossypium hirsutum L). Front Plant Sci. 2018;9:1011.
Xu P, Guo Q, Meng S, Zhang X, Xu Z, Guo W, Shen X. Genome-wide association analysis reveals genetic variations and candidate genes associated with salt tolerance related traits in Gossypium hirsutum. BMC Genomics. 2021;22(1):26.
Zheng J, Zhang Z, Gong Z, Liang Y, Sang Z, Xu Y, Li X, Wang J. Genome-wide association analysis of salt-tolerant traits in terrestrial cotton at seedling stage. Plants (Basel). 2021;11(1):97.
Zhu G, Gao W, Song X, Sun F, Hou S, Liu N, Huang Y, Zhang D, Ni Z, Chen Q, Guo W. Genome-wide association reveals genetic variation of lint yield components under salty field conditions in cotton (Gossypium hirsutum L). BMC Plant Biol. 2020;20(1):23.
Dong Y, Hu G, Yu J, Thu SW, Grover CE, Zhu S, Wendel JF. Salt-tolerance diversity in diploid and polyploid cotton (Gossypium) species. Plant J. 2020;101(5):1135–51.
Dong Y, Hu G, Grover CE, Miller ER, Zhu S, Wendel JF. Parental legacy versus regulatory innovation in salt stress responsiveness of allopolyploid cotton (Gossypium) species. Plant J. 2022;111(3):872–87.
Liang W, Ma X, Wan P, Liu L. Plant salt-tolerance mechanism: a review. Biochem Biophys Res Commun. 2018;495(1):286–91.
Zhu X, Long S, Ort D. Improving photosynthetic efficiency for greater yield. Annu Rev Plant Biol. 2010;61:235–61.
Zhang D, He S, Fu Y, Yu R, Gao X, Wang Z, Liu Z, Guo Y, Chen M. Transcriptome analysis reveals key genes in response to salinity stress during seed germination in Setaria italica. Environ Exp Bot. 2021;191:104604.
Zhang X, Liu J, Huang Y, Wu H, Hu X, Cheng B, Ma Q, Zhao Y. Comparative transcriptomics reveals the molecular mechanism of the parental lines of maize hybrid an’nong876 in response to salt stress. Int J Mol Sci. 2022;23(9):5231.
Li Q, Ai G, Shen D, Zou F, Wang J, Bai T, Chen Y, Li S, Zhang M, Jing M, Dou D. A Phytophthora capsici effector targets ACD11 binding partners that regulate ROS-mediated defense response in Arabidopsis. Mol Plant. 2019;12(4):565–81.
Martínez Y, Li X, Liu G, Bin P, Yan W, Más D, Valdivié M, Hu CA, Ren W, Yin Y. The role of methionine on metabolism, oxidative stress, and diseases. Amino Acids. 2017;49(12):2091–8.
Oliveira APDS, Melo YL, de Alencar RS, Viégas PRA, Dias GF, Ferraz RLS, Sá FVDS, Dantas Neto J, Magalhães ID, Gheyi HR, de Lacerda CF, Melo AS. Osmoregulatory and antioxidants modulation by salicylic acid and methionine in cowpea plants under the water restriction. Plants (Basel). 2023;12(6):1341.
Zhu L, Chen L, Wu C, Shan W, Cai D, Lin Z, Wei W, Chen J, Lu W, Kuang J. Methionine oxidation and reduction of the ethylene signaling component MaEIL9 are involved in banana fruit ripening. J Integr Plant Biol. 2023;65(1):150–66.
Ilangumaran G, Smith DL. Plant growth promoting rhizobacteria in amelioration of salinity stress: a systems biology perspective. Front Plant Sci. 2017;8:1768.
Peng Z, Rehman A, Li X, Jiang X, Tian C, Wang X, Li H, Wang Z, He S, Du X. Comprehensive evaluation and transcriptome analysis reveal the salt tolerance mechanism in semi-wild cotton (Gossypium purpurascens). Int J Mol Sci. 2023;24(16):12853.
Han B, Wang F, Liu Z, Chen L, Yue D, Sun W, Lin Z, Zhang X, Zhou X, Yang X. Transcriptome and metabolome profiling of interspecific CSSLs reveals general and specific mechanisms of drought resistance in cotton. Theor Appl Genet. 2022;135(10):3375–91.
Li X, Xu Y, Zhang J, Xu K, Zheng X, Luo J, Lu J. Integrative physiology and transcriptome reveal salt-tolerance differences between two licorice species: Ion transport, Casparian strip formation and flavonoids biosynthesis. BMC Plant Biol. 2024;24(1):272.
Cui C, Feng L, Zhou C, Wan H, Zhou B. Transcriptome analysis revealed that GhPP2C43-A negatively regulates salinity tolerance in an introgression line from a semi-wild upland cotton. Plant Cell Physiol. 2023;64(7):786–802.
Hayat S, Hayat Q, Alyemeni MN, Wani AS, Pichtel J, Ahmad A. Role of proline under changing environments: a review. Plant Signal Behav. 2012;7:1456–66.
Ma D, Cai J, Ma Q, Wang W, Zhao L, Li J, Su L. Comparative time-course transcriptome analysis of two contrasting alfalfa (Medicago sativa L.) genotypes reveals tolerance mechanisms to salt stress. Front Plant Sci. 2022;13:1070846.
Yu Z, Duan X, Luo L, Dai S, Ding Z, Xia G. How plant hormones mediate salt stress responses. Trends Plant Sci. 2020;25(11):1117–30.
Awan SA, Khan I, Rizwan M, Zhang X, Brestic M, Khan A, El-Sheikh MA, Alyemeni MN, Ali S, Huang L. Exogenous abscisic acid and jasmonic acid restrain polyethylene glycol-induced drought by improving the growth and antioxidative enzyme activities in pearl millet. Physiol Plant. 2021;172(2):809–19.
Peleg Z, Blumwald E. Hormone balance and abiotic stress tolerance in crop plants. Curr Opin Plant Biol. 2011;14(3):290–5.
Li Q, Xu F, Chen Z, Teng Z, Sun K, Li X, Yu J, Zhang G, Liang Y, Huang X, Du L, Qian Y, Wang Y, Chu C, Tang J. Synergistic interplay of ABA and BR signal in regulating plant growth and adaptation. Nat Plants. 2021;7(8):1108–18.
Luo X, Chen Z, Gao J, Gong Z. Abscisic acid inhibits root growth in Arabidopsis through ethylene biosynthesis. Plant J. 2014;79(1):44–55.
Peng J, Li Z, Wen X, Li W, Shi H, Yang L, Zhu H, Guo H. Salt-induced stabilization of EIN3/EIL1 confers salinity tolerance by deterring ROS accumulation in Arabidopsis. PLoS Genet. 2014;10(10):e1004664.
Wei LJ, Deng XG, Zhu T, Zheng T, Li PX, Wu JQ, Zhang DW, Lin HH. Ethylene is involved in brassinosteroids induced alternative respiratory pathway in cucumber (Cucumis sativus L.) seedlings response to abiotic stress. Front Plant Sci. 2015;6:982.
Silveira JAG, Melo ARB, Viegas RA, Oliveira JT. Salinity induced effects on nitrogen assimilation related to growth in cowpea plants. Environ Exp Bot. 2001;46:171–9.
Sousa RA, Silva J, Baccan N, Cadore. Determination of metals in bottled coconut water using an inductively coupled plasma optical emission spectrometer. J Food Compos Anal. 2005;18:399–408.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Hu Y, Chen J, Fang L, Zhang Z, Ma W, Niu Y, Ju L, Deng J, Zhao T, Lian J, Baruch K, Fang D, Liu X, Ruan YL, Rahman MU, Han J, Wang K, Wang Q, Wu H, Mei G, Zang Y, Han Z, Xu C, Shen W, Yang D, Si Z, Dai F, Zou L, Huang F, Bai Y, Zhang Y, Brodt A, Ben-Hamo H, Zhu X, Zhou B, Guan X, Zhu S, Chen X, Zhang T. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat Genet. 2019;51(4):739–48.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT, Pyysalo S, Bork P, Jensen LJ, von Mering C. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–46.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003;4:2.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆Ct method. Methods. 2001;25(4):402–8.
Acknowledgements
Not applicable.
Funding
This study was funded by China Postdoctoral Science Foundation (2024M750833), the Key Scientific and Technological Research Project of Henan Province (242102110198), the Natural Science Foundation of Jiangsu Province, China (BK20200290), the Major Science and Technology Project of Xinxiang City (22ZD001), the Science and Technology Project of Henan Province (212102110064), and the National Key Research and Development Project (2018YFD0200200).
Author information
Authors and Affiliations
Contributions
S.J. and J.L. conceived and designed the project; L.F. performed the experiments, analysed the data and wrote/revised the manuscript; Y.C., T.M., C.Z. and S.S. provided some important suggestions and supervised the experiments. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
Cite this article
Feng, L., Chen, Y., Ma, T. et al. Integrative physiology and transcriptome sequencing reveal differences between G. hirsutum and G. barbadense in response to salt stress and the identification of key salt tolerance genes. BMC Plant Biol 24, 787 (2024). https://doi.org/10.1186/s12870-024-05515-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05515-5