
Abstract
Background
Single amino acid substitutions in the Iduronate-2-sulfatase enzyme result in destabilization of the protein and cause a genetic disorder called Hunter syndrome. To gain functional insight into the mutations causing Hunter syndrome, various bioinformatics tools were employed, and special significance is given to molecular docking.
Results
In-silico tools available online for preliminary analysis including SIFT, PolyPhen 2.0, etc., were primarily employed and have identified 51 Non-synonymous Single Nucleotide Polymorphisms (ns-SNPs) as possibly deleterious. Further, modelling and energy minimization followed by Root Mean Square Deviation (RMSD) calculation has labelled 42 mutations as probably deleterious ns-SNPs. Later, trajectory analysis was performed using online tools like PSIPRED, SRide, etc., and has predicted six ns-SNPs as potentially deleterious. Additionally, docking was performed, and three candidate ns-SNPs were identified. Finally, these three ns-SNPs were confirmed to play a significant role in causing syndrome through root mean square fluctuation (RMSF) calculations.
Conclusion
From the observed results, G134E, V503D, and E521D were predicted to be candidate ns-SNPs in comparison with other in-silico tools and confirmed by RMSF calculations. Thus, the identified candidate ns-SNPs can be employed as a potential genetic marker in the early diagnosis of Hunter syndrome after clinical validation.
Similar content being viewed by others

Introduction
Hunter syndrome, a noxious and uncommon lysosomal storage disease, which is X-linked and caused by the mutation within the IDS gene [1], coding a lysosomal polypeptide of 550 amino acid length (IDS, E.C. 3.1.6.13) [2], located at chromosome Xq27.3-q28, spans 24 Kb, composed of 9 exons and belongs to the sulfatase family [3]. It is also known as type II Mucopolysaccharidosis (OMIM 309900). IDS gene codes for the significant enzyme that prevents the build-up of glycosaminoglycans (GAGs) in the lysosome.
IDS breaks-down heparan sulfate (HS) and dermatan sulfate (DS) by specifically targeting the sulfate group at the O-2 position [4]. This enzyme contains 25 amino acid residues at the amino-terminal, that acts as a signal peptide followed by 8 amino acids. These 33 amino acids are cleaved after processing and thus the functional length of the enzyme is 517 amino acids.
Activity of IDS gene is crucial for cellular maintenance [5]. Hence, defect in this gene leads to the build-up of GAGs – HS and DS in the body. It is estimated that Hunter syndrome occurs in approximately one in 170,000 male live births and prevails in individuals of all ethnicities [6, 7]. More than 500 disease-causing mutations have been identified in the IDS gene, which include single nucleotide polymorphisms (SNPs) that influence the structure and function of the enzyme greatly. Thus, SNPs can be considered as a diagnostic marker for Hunter syndrome [8].
People with Hunter syndrome show various clinical manifestations such as respiratory infections [9], sleep apnea, joint stiffness, pelvic dysplasia, hepatomegaly, umbilical and inguinal hernias, otitis, gingival hypertrophy, hyperplasia [10], cardiological and ocular manifestations, and also skin infections such as Mangolian spots and lesions [11]. Due to the non-availability of the drug, patients are advised for enzyme replacement therapy (ERT) or stem cell therapy (SCT) [12, 13].
The current study utilized a combination of bioinformatic tools to predict the structural and functional modifications of proteins and aims to identify candidate non-synonymous single nucleotide polymorphisms (ns-SNPs) in the IDS gene that may be used for specific and appropriate attuned therapies for Hunter syndrome in near future.
Methodology
Datasets and retrieval of nsSNPs
National Centre for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/snp/) is an open-source database [14], from which the non-synonymous (missense) SNPs in the IDS gene of Homo sapiens and protein sequence in FASTA format were retrieved. Recovered SNPs were subjected to look into their deleterious effect on the IDS gene by employing a set of insilico tools.
Combined prediction using preliminary tools
FASTA format of the IDS gene was given as input in tools as shown in Table 1 and ns-SNPs predicted as deleterious by all 5 tools were considered for further analysis.
Modelling and RMSD calculation
The three-dimensional structure has a momentous role in analysing the trajectory of the protein. Since no structure was available for the protein IDS, a 3-D structure was modelled through SWISS-MODEL expasy (http://swissmodel.expasy.org/) [20], validated using PROCHECK [21] and ProSA Webserver (https://prosa.services.came.sbg.ac.at/prosa.php) [22]. In due course, all mutant structures were generated using SWISS-PDB Viewer [23]. Once generated, all the structures including native were energy minimized using GROMACS, which works on l-bfgs (imited-memory Broyden-Fletcher-Goldfarb-Shanno quasi-Newtonian minimizer) [24]. Structures obtained from GROMACS were utilized for root mean square deviation (RMSD) calculation [25] using SWISS-PDB Viewer.
Trajectory analysis
IDS gene, either as a sequence or as a structure file was submitted in tools as shown in Table 2 for trajectory analysis. ns-SNPs predicted as deleterious in the maximum number of tools were selected for further analysis.
Molecular docking and visualization
Docking was performed by using Autodock Vina [32]. The results were visualized and analyzed using Ligplot+ [33].
RMSF calculation
Root mean square fluctuation (RMSF) was calculated using CABS-flex 2.0 server (http://biocomp.chem.uw.edu.pl/FCABSflex2) [34].
Effect of SNP on splicing
Splicing is the most significant post-transcriptional modification, which removes the non-coding regions and collate the exons to form the functional protein. To evaluate the influence of candidate SNPs on the splicing sites, NetGene 2 online server (https://services.healthtech.dtu.dk/service.php?NetGene2-2.42) [35] was employed. This server was provided with the DNA sequence in FASTA format as input.
Pathogenicity analysis
At last, all the candidate SNPs were employed for pathogenicity analysis to confirm and further support the prediction. This analysis was performed using DUET online server (http://biosig.unimelb.edu.au/duet/) [36].
Results and discussion
Datasets and retrieval of ns-SNPs
The IDS gene contains 5549 SNPs in total (As of 22nd September 2020). Out of these 5549 SNPs, only 289 non-synonymous SNPs (missense mutation) were retrieved from the National Centre for Biotechnology Information (NCBI) database. Missense mutation contributes 5% among total SNPs in the IDS gene (289/5549).
Combined analysis using preliminary tools
The combined analysis of five online tools had predicted 51 out of 289 ns-SNPs as deleterious in common and the individual prediction by the tools are listed in Table 3. Since the preliminary tools involve different algorithms, a combined approach may increase the precision in identifying the deleterious ns-SNPs [37]. Only the predicted 51 possibly deleterious ns-SNPs were considered for the subsequent analysis.
Modelling and RMSD calculation
A three-Dimensional structure is required for understanding the structure–function relationship of proteins, since point mutation can drastically affect protein function [38]. The 3D structure of IDS protein modelled using SWISS-MODEL was based on the template 5fql.1.A (99.81% similarity) and with the QMEAN score of -1.21.
The structure was further validated using PROCHECK and ProSA webserver. Ramachandran Plot shows the presence of 86.5% of residues within the most favourable regions, which indicates the structure obtained is in good quality [39] and the overall z-score for the protein IDS was found to be -8.14 using the ProSA server.
Since the structure obtained is a homodimer, only the Chain A was used to generate the 51 mutant structures using SWISS-PDB viewer.
Ultimately, Energy minimization was done for all structures including native using GROMACS, and RMSD values were calculated by super-imposing native and mutant structures in SWISS-PDB Viewer. Based on the RMSD values of 51 SNPs, the threshold was fixed as above 0.15 Å (> 0.15 Å). Over one-third of the mutants have RMSD value greater than 0.15, the higher the RMSD value, the higher the degree of deleterious effect [40]. Based on RMSD values, 42 SNPs were found to be probably deleterious and are enlisted in Table 4.
Trajectory analysis
Only the possibly deleterious ns-SNPs (42 ns-SNPs) were analysed using trajectory tools. The analysis had predicted 6 ns-SNPs (G134E, P358T, R468W, M488I, V503D, and E521D) as potentially deleterious in common by maximum number of tools (Table 5). Multiple approaches were employed for trajectory analysis using various tools, to get more accurate results. Among these ns-SNPs five SNPs (G134E, P358T, M488I, V503D, and E521D) have variations in their secondary structures after mutation and all these 6 SNPs have modified stability residues in comparison with the native structures. The conservation score also varies significantly along with the Relative Solvent Accessibility values. Analogously, a study has been communicated, which also employs these properties for the prediction of most deleterious mutation in HBA1 gene [39].
Molecular docking and visualization
Protein–Ligand Docking Analysis demonstrated that the mutant structures bind to the ligand in a slightly different orientation compared to the native protein. Three ligands were used for docking within which two were the enzyme’s own substrates (dermatan sulfate, DS and heparan sulfate, HS) and one from a plant source (Luteolin Sulfate -LS). LS was selected as a ligand because of its abundant availability [41] and its structural similarity with HS and DS. The binding energy of the ligands with mutants were compared with native protein.
InterPro server [42] and literature survey [5] identified D45, D46, C84, R88, K135, F137, H138, H229, D334, H335, and K347 (11 residues) as active site residues, and site-specific docking was performed using Autodock Vina. From the results of docking, it is obvious that binding energy differs drastically in the case of G134E, V503D, and E521D (especially for the ligand—Heparan Sulfate) (Table 6). Binding energies for remaining residues vary very slightly. Thus, HS was hypothesised to play a crucial role in causing Hunter’s syndrome and these three ns-SNPs were considered as candidate ns-SNPs. Binding energy of these mutant proteins with HS in the presence of LS were calculated (Table 6). From the results, it is observed that binding of LS with the protein alters the protein structure and increases the affinity for the ligand. Thus, LS may act as a potential molecule in treating Hunter Syndrome.
The interaction between native and mutant proteins (G134E, V503D, and E521D) with the ligand Heparan Sulfate were visualized and bonds formed in native and mutants were observed using ligplot+ (Fig. 1). Ligplot+ analysis shows the variations in bonds formed by mutant proteins with ligands, compared with native protein. The mutation could also be the rational for these variations [43] and it supports the conclusions obtained from all the previous analyses. These predictions are in accordance with the deviation in RMSD values, as predicted earlier.

Pictorial representation of the analysis using Ligplot+ tool. a Native-HS complex, b G134E-HS complex, c V503D-HS complex and d E521D-HS complex
RMSF calculation
RMSF calculation done using the CABS-flex server, confirms G134E, V503D, and E521D as candidate ns-SNPs with variations in RMSF values (Table 7). Obtained result revealed that the values for mutants were lower when compared with native structure. This confirms the compressed nature of protein structure which in turn can affect the protein function [44].
Effect of SNP on splicing
Netgene 2 employs an artificial neural network that predicts the splice site location. The DNA sequence in FASTA format for native and all three identified candidate SNPs were given as inputs and splice sites were predicted. The results of the candidate SNPs splice site prediction were compared with the native prediction. As a result, we found that these SNPs do not interfere with the splicing mechanism of the IDS gene.
Pathogenicity analysis
Pathogenicity analysis was performed using DUET web server. It is an online server used to analyse the pathogenic effects of missense mutations in proteins. This server provides the integrative result by combining two independent approaches, namely mCSM and SDM. The results of this analysis are provided in Table 8, which predicts that all the three identified candidate SNPs will result in the destabilization of protein. This further supports the identification of G134E, V503D, and E521D as candidate SNPs.
Conclusion
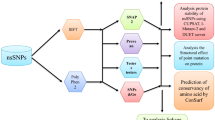
Analysis using various insilico tools predicts the influence of ns-SNPs on the structure and function of protein IDS. Out of 289 ns-SNPs, G134E, V503D and E521D with SNP IDs—rs193302910, rs398123248, and rs1602725543, respectively, were predicted as candidate ns-SNPs. These ns-SNPs may alter the structure of the protein and interfere with the functions. The result summary of the complete workflow is provided in Fig. 2 and the structure of the mutant is compared with the native structure using PyMol [45] in Fig. 3. The presence of these ns-SNPs may inactivate the enzyme IDS and results in accumulation of GAGs, ultimately leading to Hunter syndrome. These ns-SNPs can be considered for clinical confirmatory studies for understanding the exact mechanism and pathology of mutations, before used for diagnostics.

Result summary of the entire work-flow

Comparison of native (left) and mutant (right) structure using PyMol. a G134E, b V503D and c V521D
Availability of data and materials
All data analysed during the current study are available in the National Centre for Biotechnology Information (NCBI) repository, https://www.ncbi.nlm.nih.gov/.
References
Yatziv S, Erickson RP, Epstein CJ (1977) Mild and severe Hunter syndrome (MPS II) within the same sibships. Clin Genet 11:319–326. https://doi.org/10.1111/j.1399-0004.1977.tb01323.x
Galvis J, González J, Uribe A, Velasco H (2015) Deep genotyping of the IDS Gene in Colombian Patients with Hunter Syndrome. JIMD Rep 19:101–109. https://doi.org/10.1007/8904_2014_376
Wilson PJ, Morris CP, Anson DS, Occhiodoro T, Bielicki J, Clements PR et al (1990) Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci U S A 87:8531–8535. https://doi.org/10.1073/pnas.87.21.8531
Shaklee P, Glaser J, Conrad H (1985) A sulfatase specific for glucuronic acid 2-sulfate residues in glycosaminoglycans. J Biol Chem 260:9146–9149
Demydchuk M, Hill C, Zhou A, Bunkoczi G, Stein PE, Marchesan D et al (2017) Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat Commun 8:15786. https://doi.org/10.1038/ncomms15786
Young ID, Harper PS (1982) Incidence of Hunter’s syndrome. Hum Genet 60:391–392. https://doi.org/10.1007/BF00569230
Nelson J, Crowhurst J, Carey B, Greed L (2003) Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A 123A:310–313. https://doi.org/10.1002/ajmg.a.20314
Schumacher RG, Brzezinska R, Schulze-Frenking G, Pitz S (2008) Sonographic ocular findings in patients with mucopolysaccharidoses I, II and VI. Pediatr Radiol 38:543–550. https://doi.org/10.1007/s00247-008-0788-y
Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Muñoz V, Muenzer J (2008) Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 121:e377–e386. https://doi.org/10.1542/peds.2007-1350
Sanjurjo-Crespo P (2007) Mucopolisacaridosis de tipo II: aspectos clínicos (Clinical aspects of mucopolysaccharidosis type II). Rev Neurol. 44 Suppl 1:S3–S6 (Spanish)
Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N et al (2008) Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr 167:267–277. https://doi.org/10.1007/s00431-007-0635-4
Giugliani R, Villarreal MLS, Valdez CAA, Hawilou AM, Guelbert N, Garzon LNC et al (2014) Guidelines for diagnosis and treatment of Hunter Syndrome for clinicians in Latin America. Genet Mol Biol 37:315–329. https://doi.org/10.1590/s1415-47572014000300003
Bradley L, Haddow H, Palomaki G (2017) Treatment of mucopolysaccharidosis type II (Hunter syndrome): results from a systematic evidence review. Genet Med 19:1187–1201. https://doi.org/10.1038/gim.2017.30
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) dbSNP: the NCBI database of genetic variation. Nucl Acids Res 29:308–311. https://doi.org/10.1093/nar/29.1.308
Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucl Acids Res 40(Web Server issue):W452-7. https://doi.org/10.1093/nar/gks539
Adzhubei IA, Schmidt S, Peshkin L, Ramensky V, Gerasimova A, Bork P, Kondrashov A, Sunyaev SA (2010) Method and server for predicting damaging missense mutations. Nat Methods 7:248–249. https://doi.org/10.1038/nmeth0410-248
Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucl Acids Res 47:D419–D426. https://doi.org/10.1093/nar/gky1038
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucl Acids Res 33(Web Server issue):W306–W310. https://doi.org/10.1093/nar/gki375.10.1093/nar/gki375
López-Ferrando V, Gazzo A, de la Cruz X, Orozco M, Gelpí JL (2017) PMut: a web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucl Acids Res 45:W222–W228. https://doi.org/10.1093/nar/gkx313
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucl Acids Res 46:W296-303. https://doi.org/10.1093/nar/gky427
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291. https://doi.org/10.1107/S0021889892009944
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl Acids Res 35:W407–W410. https://doi.org/10.1093/nar/gkm290
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. https://doi.org/10.1002/elps.1150181505
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Coutsias EA, Seok C, Dill KA (2004) Using quaternions to calculate RMSD. J Comput Chem 25:1849–1857. https://doi.org/10.1002/jcc.20110
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. https://doi.org/10.1006/jmbi.1999.3091
Magyar C, Gromiha MM, Pujadas G, Tusnády GE, Simon I (2005) SRide: a server for identifying stabilizing residues in proteins. Nucl Acids Res 33(Web Server issue):W303–W305. https://doi.org/10.1093/nar/gki409
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucl Acids Res 44:W344–W350. https://doi.org/10.1093/nar/gkw408
Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Sønderby CK, Sommer MOA, Winther O, Nielsen M, Petersen B, Marcatili P (2019) NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins Struct Funct Bioinform 87:520–7. https://doi.org/10.1002/prot.25674
de Brevern AG, Bornot A, Craveur P, Etchebest C, Gelly JC (2012) PredyFlexy: flexibility and local structure prediction from sequence. Nucl Acids Res 40(Web Server issue):W317-22. https://doi.org/10.1093/nar/gks482
Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJE (2019) Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J Mol Biol 431:2197–2212. https://doi.org/10.1016/j.jmb.2019.04.009
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Prot Eng 8:127–134. https://doi.org/10.1093/protein/8.2.127
Kuriata A, Gierut AM, Oleniecki T, Ciemny MP, Kolinski A, Kurcinski M, Kmiecik S (2018) CABS-flex 2.0: a web server for fast simulations of flexibility of protein structures. Nucl Acids Res. 46:W338-43. https://doi.org/10.1093/nar/gky356
Brunak S, Engelbrecht J, Knudsen S (1991) Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 220:49–65. https://doi.org/10.1016/0022-2836(91)90380-O
Pires DE, Ascher DB, Blundell TL (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucl Acids Res 42(W1):W314–W319. https://doi.org/10.1093/nar/gku411
Meléndez-Aranda L, Jaloma-Cruz AR, Pastor N, Romero-Prado MMdeJ (2019) In silico analysis of missense mutations in exons 1–5 of the F9 gene that cause hemophilia B. BMC Bioinform 20:363. https://doi.org/10.1186/s12859-019-2919-x
Pandey S, Dhusia K, Katara P, Singh S, Gautam B (2019) An in-silico analysis of deleterious single nucleotide polymorphisms and molecular dynamics simulation of disease linked mutations in genes responsible for neurodegenerative disorder. J Biomol Struct Dyn 1:22. https://doi.org/10.1080/07391102.2019.1682047
AbdulAzeez S, Borgio JF (2016) In-silico computing of the most deleterious nsSNPs in HBA1 gene. PLoS ONE 11:e0147702. https://doi.org/10.1371/journal.pone.0147702
Chandrasekaran G, Hwang EC, Kang TW, Kwon DD, Park K, Lee JJ, Lakshmanan VK (2017) In silico analysis of the deleterious nsSNPs (missense) in the homeobox domain of human HOXB13 gene responsible for hereditary prostate cancer. Chem Biol Drug Des 90:188–199. https://doi.org/10.1111/cbdd.12938
Manzoor MF, Ahmad N, Manzoor A, Kalsoom A (2017) Food based phytochemical luteolin their derivatives, sources and medicinal benefits. IJALS 3:195–207. https://doi.org/10.22573/spg.ijals.017.s12200084
Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M et al (2001) The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucl Acids Res 29:37–40. https://doi.org/10.1093/nar/29.1.37
Sunkar S, Neeharika D (2020) CYP2R1 and CYP27A1 genes: An in silico approach to identify the deleterious mutations, impact on structure and their differential expression in disease conditions. Genomics 112:3677–3686. https://doi.org/10.1016/j.ygeno.2020.04.017
Pandey S, Dhusia K, Katara P, Singh S, Gautam B (2019) An in-silico analysis of deleterious single nucleotide polymorphisms and molecular dynamics simulation of disease linked mutations in genes responsible for neurodegenerative disorder. J Biomol Struct Dyn 38:4259–4272. https://doi.org/10.1080/07391102.2019.1682047
Yuan S, Chan HCS, Hu Z (2017) Using PyMOL as a platform for computational drug design. Wiley Interdiscip Rev Comput Mol Sci 7:e1298. https://doi.org/10.1002/wcms.1298
Acknowledgements
The authors are thankful to CSIR-NEIST for providing an opportunity to carry out this work. The authors thank the Researchers Supporting Project Number (RSP-2021/293) for financial support and King Saud University, Riyadh, Saudi Arabia.
Funding
This study was funded by King Saud University (RSP-2021/293).
Author information
Authors and Affiliations
Contributions
AS: methodology, investigation, resources, data curation, writing–original draft. YD: project administration. WHA-Q: funding acquisition. MK: project administration. JR: project administration. AC: project administration. ES: investigation, resources, writing–review and editing. APV: Investigation, resources. BMS: investigation, resources. MQ: investigation, resources. SRA: conceptualization, methodology, validation, writing–review and editing, supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sivakumar, A., Dinakarkumar, Y., Al-Qahtani, W.H. et al. In silico profiling of non-synonymous SNPs in IDS gene for early diagnosis of Hunter syndrome. Egypt J Med Hum Genet 23, 53 (2022). https://doi.org/10.1186/s43042-022-00271-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00271-3